Urticaria and mimickers of urticaria
 Jie Shen Fok1,2,3*†
Jie Shen Fok1,2,3*†  Constance H. Katelaris4,5,†
Constance H. Katelaris4,5,†
- 1Department of Respiratory Medicine and General Medicine, Box Hill Hospital, Eastern Health, Melbourne, VIC, Australia
- 2Monash Lung, Sleep and Allergy/Immunology, Monash Medical Centre, Melbourne, VIC, Australia
- 3Eastern Health Clinical School, Monash University, Melbourne, VIC, Australia
- 4Department of Medicine, Immunology and Allergy Unit, Campbelltown Hospital, Sydney, NSW, Australia
- 5School of Medicine, Western Sydney University, Sydney, NSW, Australia
Urticaria is a common skin condition encountered across various specialties in medicine, especially in dermatology and allergy/immunology practice. It has a heterogeneous presentation hence it is unsurprising that many skin conditions may be confused with urticaria. Urticaria may present as acute or chronic urticaria, the latter can be further categorised into chronic spontaneous and chronic inducible. In this article, we explore, explain, and summarise various skin lesions that are considered mimickers of urticaria, to promote understanding of each of the conditions highlighted, improve recognition, and reduce misdiagnosis.
Introduction
Urticaria is a common presentation in general practice and a common referral to clinicians including dermatologists, allergists, and immunologists. It is an inflammatory skin disorder characterised by wheals, hives, and often intractable itch, with an estimated lifetime prevalence of 20% globally (1). A typical urticarial wheal has a circumscribed superficial central swelling that may present with various sizes and shapes, surrounded by erythema, often with rapid appearance, and may appear on different locations of the body, including the face, trunk, and limbs (Figure 1). It has a fleeting nature, with resolution to its normal appearance within 24 h. The intensity of the itch is known to fluctuate during the course of its appearance. Angioedema, a swelling with similar pathogenesis, involves the submucosal, lower dermis, and subcutaneous tissues. It may occur suddenly, with pronounced erythematous or skin-coloured swelling (2). Resolution of angioedema is slower than urticarial wheal and may take up to days (e.g., 48–72 h). Urticaria and angioedema may occur alone or together, in both paediatric and adult populations.
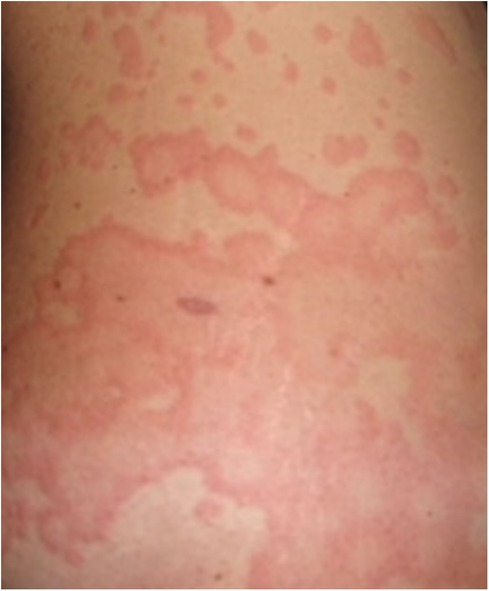
Figure 1. An urticarial wheal.
Atypical urticarial lesions may appear more indurated, more discoloured, dusky, or bruised in appearance, and last longer. The lesions may heal with an eventual hyperpigmented mark. Instead of pruritic, they are painful. Angioedema is typically absent. Systemic features may coexist, including arthralgia, fever, and malaise.
Urticaria can be categorised as acute or chronic. Acute urticaria is recurrent flares of hives lasting less than 6 weeks. It occurs most often in children, in the setting of upper respiratory viral infection in most cases (3). In other settings, acute urticaria presents as part of the spectrum of an acute allergic reaction, caused by drug allergy, food allergy, or Hymenoptera sting allergy. Commonly implicated drug causes in real-life practice include penicillin and non-steroidal anti-inflammatory drugs. Acute urticaria and angioedema are also manifestations of anaphylaxis, with progression to respiratory, cardiovascular, or gastrointestinal involvement following exposure to an allergen that triggers a generalised immune response with the release of various chemical mediators.
Chronic urticaria is defined as the appearance of hives occurring on most days of the week lasting for more than 6 weeks. A majority of chronic urticaria cases are chronic spontaneous urticaria (CSU), with two distinctive subtypes being recognised: autoallergic CSU (type 1 autoimmunity) with IgE auto-antibody involvement and autoimmune CSU (type IIb autoimmunity) with IgG auto-antibody involvement (4, 5). Chronic inducible urticaria (CIndU), on the other hand, is less prevalent than CSU and accounts for approximately 13% of chronic urticaria cases. It is also known as physical urticaria, aptly described because of a physical trigger that induces the appearance of wheals. The most prevalent CIndU types are symptomatic dermographism, cold urticaria, and cholinergic urticaria.
There is, of course, a list of differential diagnoses to be considered for CSU. These include CIndU, Schnitzler syndrome, cryopyrin-associated periodic syndrome, Still's disease, and urticarial vasculitis, with specific questions and aspects of physical examination that could guide further evaluation before a final diagnosis is determined (6).
There are also several skin lesions with morphology that resemble an urticarial lesion, which may pose challenges to even the most experienced clinician. Here, we discuss a list of cutaneous lesions that may be regarded as mimickers of urticaria to improve understanding of a common presenting problem in dermatology or allergy/immunology practice.
Urticaria pigmentosa
Mastocytosis is a clonal disorder characterised by abnormal mast cells in various organs, including the skin, bone marrow, liver, and gastrointestinal tract. It is the result of abnormal expansion and focal accumulation of neoplastic mast cells driven by a gain-of-function mutation in KIT, a membrane protein that keeps growth and activation of mast cells in check. The most common mutation described is KIT D816V, though there are other less commonly described mutations. The spectrum of mastocytosis presentation is heterogeneous, ranging from cutaneous to systemic involvement. Cutaneous mastocytosis (CM) is largely benign, whereas systemic mastocytosis often has serious implications, particularly increased risks for anaphylaxis and lymphoproliferation.
CM is further categorised into maculopapular cutaneous mastocytosis (also known as urticaria pigmentosa), diffuse cutaneous mastocytosis, and mastocytoma of the skin. Most cases of CM occur in childhood, and in most cases, lesions usually regress by puberty (7). In most cases, bone marrow examination is not routinely required unless there are features that suggest systemic mastocytosis or lymphoproliferative neoplasm (8).
Urticaria pigmentosa was first reported in 1,869 in a 2-year-old child who presented with urticaria with a brownish stain (9). It was not until 1,878 that the term urticaria pigmentosa was coined (10). It is by far the most common form of CM, characterised by macules and papules, hence the aptly named maculopapular CM. It may have a brownish discolouration, with two distinctive variants recognised so far. The monomorphic variant is seen in both children and adults. The polymorphic variant is usually observed in childhood and has a tendency to disappear by puberty. A key feature of UP is a positive Darier's sign, which is considered a pathognomonic sign for CM, named after the 19th-century French dermatologist Ferdinand-Jean Darier. Darier's sign is positive when a CM lesion is gently rubbed or stroked, resulting in local itch and whealing within a few minutes. It is postulated that the phenomenon is caused by the degranulation of dermal mast cells with the release of mediators.
Skin biopsy is characterised by increased mast cell numbers in the papillary dermis and around blood vessels. Immunochemistry staining with mast cell tryptase antibody helps detect mast cell infiltrates. Using CD117 immunostaining, the mast cell distribution pattern and the percentage of mast cells in the inflammatory infiltrates can be determined (11). The role of skin biopsy is also important in prognosticating UP. The monomorphic variant is associated with sparing of the papillary dermis from mast cell infiltration. On the contrary, mast cell density in the papillary dermis has been found highest in the polymorphic variant and diffuse CM (12).
Erythema multiforme
Erythema multiforme (EM) is a cutaneous and mucosal hypersensitivity reaction triggered by a stimulus. Infectious aetiology accounts for most cases of EM, with herpes simplex virus (HSV) being the most common cause in adults (13). Mycoplasma and Epstein-Barr virus are other commonly implicating organisms. Drug causes account for the remaining cases in clinical practice, including beta-lactam antibiotics, sulphonamide antibiotics, and anti-convulsants such as phenobarbital.
The emergence of the novel Coronavirus disease-19 (COVID-19) infection has seen cases of EM being reported as a cutaneous complication, either to the infection itself or to the vaccine (14, 15). EM due to COVID-19 vaccination occurred most often after the first dose, most commonly with mRNA vaccines in one study (15). In another study, EM was more commonly reported in COVID-19 infection compared to COVID-19 immunisation (16). Small numbers of cases make it challenging to establish a causal relationship. Numerous other vaccinations including combined mump, measles, and rubella vaccine, combined diphtheria, tetanus, and pertussis vaccine, varicella vaccine, pneumococcal vaccine, smallpox vaccine, and influenza vaccine have all been reported to cause EM in the Vaccine Adverse Event Reporting System (17).
EM has an acute presentation. It starts with a painful eruption, often an erythematous papule, that eventually progresses to form a target lesion. Its resemblance to urticaria is related to its lighter oedematous appearance surrounded by a peripheral erythematous border (Figure 2). One of the main characteristics of the target lesion is its central dusky area which is not seen in urticaria. The central area may not necessarily blister. In fact, the hallmark of the target lesion is its three distinct, concentric zones with colour change (18, 19). On the mucous membrane, the lesion may form a blister and erode, forming a crust as it heals. It may involve mucous membranes of the oral cavity, eyes, or genitals. Ocular involvement may pose serious complications, including conjunctival scarring, keratitis, and visual impairment. Recurrent cases in the paediatric cohort are commonly linked to HSV (20).
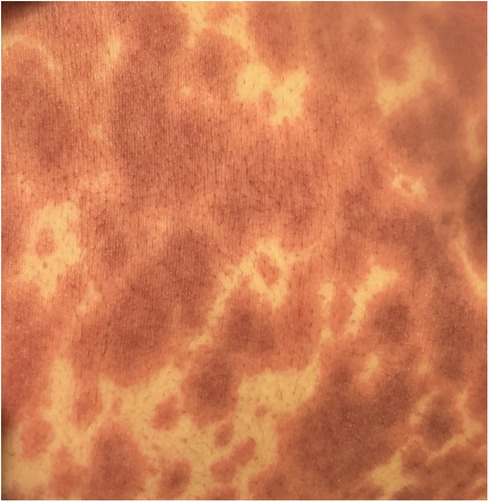
Figure 2. Erythema multiforme in a subject with herpes simplex virus infection.
Diagnosis of EM is based on clinical examination of its characteristic morphology. It is supported by investigations that further evaluate an infective aetiology, including full blood count, inflammatory markers, polymerase chain reaction (PCR) test for HSV from a concurrent skin lesion (e.g., a cold sore), nasal PCR test for Mycoplasma pneumoniae, and chest x-ray looking for pulmonary infiltrates. A skin biopsy should be performed if in doubt. Characteristic histopathological findings include keratinocyte necrosis, particularly in the centre of the targetoid lesion (21). Necrosis of the whole dermis may be seen in severe cases. Direct immunofluorescence is negative.
Urticaria vasculitis
Urticarial vasculitis (UV) is characterised by long-standing urticarial rashes and histopathologic findings of leukocytoclastic vasculitis (22). The lesion has an inflamed and brightly erythematous wheal that resembles urticaria, with the additional feature of vessel inflammation. UV being a systemic disease may involve internal organs. An array of conditions, ranging from autoimmune and infective to paraneoplastic, have been associated with UV. Autoimmune diseases [e.g., Systemic Lupus Erythematosus (SLE) and Sjögren's Syndrome], haematological diseases (e.g., lymphoproliferative diseases and monoclonal gammopathy) (23, 24), infections (e.g., Streptococcus and viral hepatitis) (25), and drugs (e.g., antibiotics) can all cause UV, though in the majority of cases, the cause is unknown.
There are two types of UV, known as normocomplementaemic UV and hypocomplementaemic UV (McDuffie syndrome). Each type has unique pathophysiology, clinical manifestations, and prognosis. UV is considered a complement-mediated disease that is considered a type-3 hypersensitivity reaction. The pathophysiology is a complex interplay between the interaction of antibodies with antigens resulting in complement cascade activation and production of C3a, C5a, and C5b-9 via the classical pathway (26, 27). Systemic involvement is more commonly seen in HUV than in NUV (28, 29).
The unique role of anti-C1q autoantibodies in hypocomplementaemic UV (HUV) is explained by the activation of complement cascade by autoantibodies targeting the collagen-like region of C1q (30). The pathogenic role of anti-C1q autoantibodies in immune complex-mediated renal disease is supported by previous studies, with further observation of level rise in anti-C1q antibodies prior to a relapse of lupus nephritis, as early as six months in advance (31, 32). Anti-C1q antibodies were found to specifically target C1q bound on cells undergoing apoptosis in a previous study involving patients with SLE, indicating that early apoptotic cells are a major target for the autoimmune response (33). Additionally, it has been postulated that pulmonary insult in HUV is a result of anti-C1q antibodies cross-reacting with pulmonary alveolar surfactant apoprotein that contains C1q collagen-like proteins, resulting in chronic obstructive airway disease (34).
The hallmark symptom of an urticarial vasculitis lesion is a painful and burning sensation, in contrast to the wheals of acute or chronic urticaria which are typically pruritic. It is a common mimic of urticaria, however, additional clues to UV include a bruised appearance, longer duration of lesions, often for days (in contrast to acute or chronic urticaria lesions that tend to resolve within a few hours), and healing with residual post-inflammatory ecchymotic hyperpigmented marks.
Constitutional symptoms, such as malaise and fever, as well as internal organ involvement, may occur. Arthralgia may be present in approximately 50% of cases (35). Any joints may be affected, commonly the joints of the upper and lower limbs, including the hands and feet. Renal involvement is not uncommon, necessitating further workup to exclude haematuria and proteinuria, and if found, further characterisation of intrinsic kidney involvement may be warranted, such as renal biopsy. Well-described associations include membranoproliferative, mesangioproliferative, and crescentic glomerulonephritis (36, 37). Pulmonary involvement may be present in UV, particularly obstructive airway diseases such as asthma and chronic obstructive airway disease. Vasculitis may also involve the gastrointestinal tract and eyes.
Clinical assessment for UV should always include a systemic review given internal organ involvement, especially in HUV. Physical examination may reveal tell-tale signs of UV, especially from the cutaneous aspect. Thorough investigations including full blood count, inflammatory markers, serum complements, renal function, and liver enzymes are considered basic. Further exploration into a potential autoimmune, infective, or neoplastic aetiology should be undertaken, including antinuclear antibodies, extractable nuclear antigens, anti-double-stranded DNA antibodies, rheumatoid factor, anti-citrullinated protein antibodies, cryoglobulins, anti-neutrophil cytoplasmic antibodies, viral hepatitis serology, anti-streptolysin O titres, full blood picture, lymphocyte immunophenotyping, serum protein electrophoresis, serum free light chain, tumour markers, and urine Bence-Jones protein. Urine analysis looking for the presence of protein, blood, or casts, should be performed. Renal biopsy may be required, especially to confirm or establish the type of glomerulonephritis. In the setting of pulmonary involvement, chest x-ray, chest computed tomography, lung function tests, and blood gas measurement are all useful. With musculoskeletal involvement, a skeletal survey is recommended.
Skin biopsy demonstrates features suggestive of leukocytoclastic vasculitis. The key feature is neutrophil predominance, in contrast to non-vasculitic urticarial samples, which may show mostly the presence of eosinophils. A novel histopathological diagnostic scoring system was established recently to distinguish UV from CSU. A combined quantitative assessment of three key features of leukocytoclasia, fibrin deposits, and extravasated erythrocytes convincingly distinguishes UV from CSU in skin histopathology reading (38). This scoring system serves as a useful tool to reduce the chance of misdiagnosis of UV.
Erythema marginatum
Erythema marginatum (EM) is a reticular, serpiginous, erythematous lesion that may be mistaken for urticaria. Unlike urticaria, EM is not pruritic and is less widespread. EM was first described by Dinckelacker in 1882. EM is commonly associated with Hereditary Angioedema (HAE), where it may occur alone, or together with fatigue and malaise, as a prodromal symptom experienced by patients with HAE prior to an imminent acute attack of angioedema. In certain parts of the world, including Central and Northern Australia and Southeast Asia where rheumatic fever is endemic, EM is one of the major criteria for this infection caused by Streptococcus.
EM may occur on any part of the body, including the trunk and limbs. Given its morphology mimicking urticaria, it is unsurprising that patients with HAE presenting with EM and angioedema potentially may be misdiagnosed as having an allergy. Recent studies have highlighted the rate of misdiagnosis or delayed diagnosis of HAE given misinterpretation of EM as urticaria. Diagnostic delay for 2 years in HAE patients with EM had previously been found (39).
Prodromal symptoms occur hours to days before an acute attack in HAE, with almost two-thirds in a large German population study experiencing the symptoms within 6 h before an attack (40). The syndrome consists of fatigue, malaise, short temper, and EM. There was a significant association between EM and delayed diagnosis of HAE.
In the setting of HAE, EM may occur in HAE with C1 esterase inhibitor (C1-INH) deficiency as well as with normal C1-INH levels. This was observed in a Japanese survey on prodromal symptoms in patients with HAE. The same study reported EM distribution on the forearm as the predominant site, followed by the abdomen, upper arm, and precordium (41).
HAE is an inherited condition with or without C1-INH deficiency. In type 1 HAE, both C1-INH function and antigenic levels are low. In type 2 HAE, C1-INH function is low, whereas C1-INH antigenic level may be normal or elevated. Both types are caused by SERPING1 gene mutations (42). Known genetic mutations in HAE with normal C1-INH are factor XII, plasminogen, angiopoietin-1, kininogen-1, myoferlin, and heparan sulfate glucosamine 3-o-sulfotransferase 6 (43–48). Each of these mutations results in distinctive pathophysiology in HAE with normal C1INH. For example, in HAE with angiopoietin-1 the mutation alters the ability of the angiopoietin-1 gene to modulate endothelial permeability induced by vascular endothelial growth factor, resulting in vascular leakage (45).
Urticarial dermatitis
Urticarial dermatitis (UD) is an intensely pruritic skin eruption that may exist in papules and plaques resembling urticaria. Unlike urticaria, which has a rapid resolution, UD often lasts longer than 24 h. It is recognised that UD may persist for days to even weeks (49). Common distribution includes the trunk and extremities, with the palms and soles typically spared. Like many eczematous dermatoses, chronic scratching may result in lichenification.
Skin biopsy is helpful when certain features unique to other dermatoses are absent. Overall, non-specific findings are seen on skin biopsy taken from a UD lesion. These include epidermal oedema with perivascular inflammatory (often lymphocytic) infiltrate, with eosinophils present in the papillary and upper reticular dermis (50). Dermal hypersensitivity reaction pattern is a common descriptive phrase. Importantly, prominent spongiosis when present, points towards a diagnosis of eczematous dermatoses.
Bullous pemphigoid
Bullous pemphigoid (BP) is an autoimmune, heterogeneous, blistering skin disease affecting mostly older adults. It may involve both the skin and mucous membranes. BP is intensely pruritic in the prodromal phase, characterised by non-bullous lesion eruption, often in the form of excoriated, urticarial, or eczematous lesions (51, 52). As the disease progresses, tense blisters form on the urticarial base and become more widespread. Scarring is not usually a feature of BP as the bullae rupture and heal. Though various mucosal tissues may be involved, oral mucosa is the most frequently affected mucosal surface (53).
Skin biopsy of lesional tissue and direct immunofluorescent (DIF) staining of the perilesional tissue specimen are key investigations. Key histopathological findings include subepithelial blister formation and eosinophilic spongiosis (52). Skin biopsy interpretation of an early non-bullous lesion can be challenging because of non-specific findings (54). DIF typically demonstrates staining of linear IgG and linear C3 along the basement membrane zone in most cases. Indirect immunofluorescent (IIF) testing is utilised to detect circulating IgG antibodies along the basement membrane zone. Enzyme-linked immunosorbent assay (ELISA) detects circulating IgE antibodies against BP180 and BP230, which are pathogenic autoantigens in BP (55–57). In particular, IgE anti-BP230 antibodies have been shown to play an important role in local eosinophil accumulation in skin lesions.
Polymorphic eruption of pregnancy
*also known as pruritic urticarial papules and plaques of pregnancy (PUPPP)
Pruritic urticarial papules and plaques of pregnancy (PUPP) is a benign inflammatory disorder that is the most common dermatosis in pregnancy, appearing most commonly in the last weeks of pregnancy. Itchy small papules and plaques start in stretch marks and spare the periumbilical area. The eruption generally spreads to the trunk and limbs but rarely involves the face. There is no mucosal involvement. It resolves rapidly postpartum, leaving no pigmentation or scarring.
The cause of the condition is unknown but several factors are considered, including rapid abdominal distension, hormonal changes, and placental and foetal factors. One theory suggests that connective tissue damage within striae is a factor.
It is seen in primigravidas primarily and does not typically recur in later pregnancies (58). The incidence is higher in twin (3%–16%) and triplet (14%–17%) pregnancies compared to single pregnancies (0.5%). There are no familial links or underlying autoimmune diatheses identified.
Biopsy specimens from lesions show non-specific features and direct immunofluorescence is usually negative. Routine laboratory tests are within normal limits.
Symptomatic treatment is usually all that is required with topical steroids and antihistamines commonly prescribed. If severe, causing sleep disturbance, a very short course of oral corticosteroids may be considered.
Apart from the discomfort, there are no negative consequences; pregnancy and maternal prognosis are not affected (58).
Characteristic features and investigations of each of the conditions are summarised in Table 1.
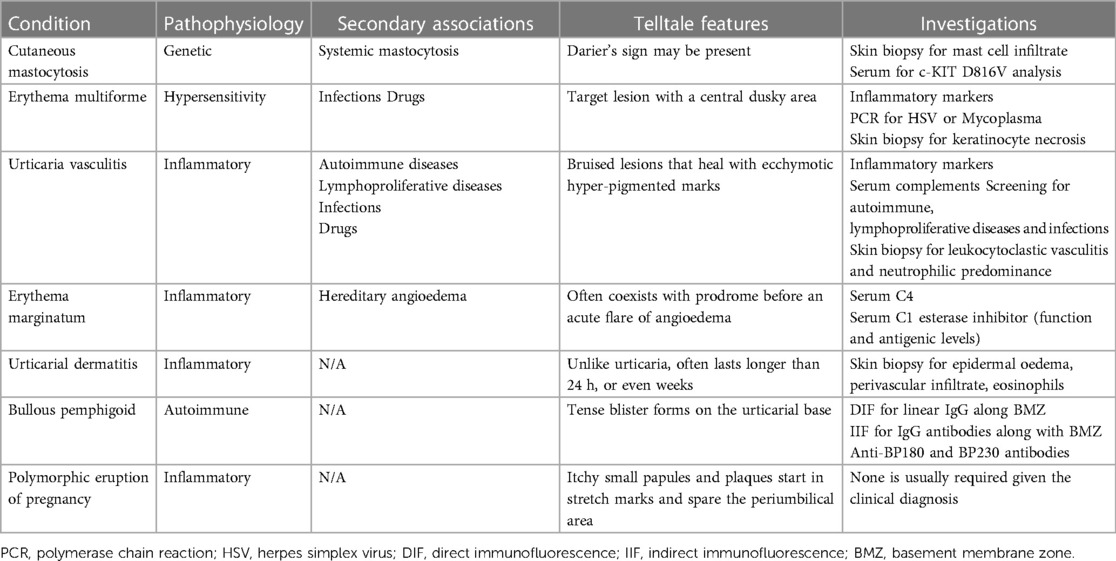
Table 1. Summary of mimickers of urticaria.
Conclusion
There are many dermatological conditions that resemble the appearance of urticaria, making diagnosis sometimes challenging. With an understanding of underlying disease associations and pathology, together with a recognition of pathognomonic features, a diagnosis can be established with the aid of relevant investigations.
Author contributions
JF and CK: Conceptualization, Writing – review and editing.
Conflict of interest
CK has received honoraria for presentations and fees for advisory board participation from Sanofi, CSL Behring, Takeda, GSK, Novartis, Abvvie, AstraZeneca, KalVista. She has received recent institutional funding for clinical trials from Sanofi, CSL Behring, Takeda. JF has received honoraria for presentations and fees for advisory board participation from CSL Behring, Takeda, Novartis, Menarini, Viatris.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kolkhir P, Giménez-Arnau A, Kulthanan K, Peter J, Metz M, Maurer M. Urticaria. Nat Rev Dis Primers. (2022) 8(1):61. doi: 10.1038/s41572-022-00389-z
2. Zuberbier T, Abdul Latiff AH, Abuzakouk M, Aquilina S, Asero R, Baker D, et al. The international EAACI/GA2LEN/EuroGuiDerm/APAAACI guideline for the definition, classification, diagnosis, and management of urticaria. Allergy. (2022) 77(3):734–66. doi: 10.1111/all.15090
3. Ensina LF, Brandão LS, Neto HC, Ben-Shoshan M. Urticaria and angioedema in children and adolescents: diagnostic challenge. Allergol Immunopathol (Madrid). (2022) 50(S Pt 1):17–29. doi: 10.15586/aei.v50iSP1.538
4. Schoepke N, Asero R, Ellrich A, Ferrer M, Giménez-Arnau A, Grattan C, et al. Biomarkers and clinical characteristics of autoimmune chronic spontaneous urticaria: results of the PURIST study. Allergy. (2019) 74(12):2427–36. doi: 10.1111/all.13949
5. Schmetzer O, Lakin E, Topal FA, Preusse P, Freier D, Church MK, et al. IL-24 is a common and specific autoantigen of IgE in patients with chronic spontaneous urticaria. J Allergy Clin Immunol. (2018) 142(3):876–82. doi: 10.1016/j.jaci.2017.10.035
6. Metz M, Altrichter S, Buttgereit T, Fluhr JW, Fok JS, Hawro T, et al. The diagnostic workup in chronic spontaneous urticaria – what to test and why. J Allergy Clin Immunol Pract. (2021) 9(6):2274–83. doi: 10.1016/j.jaip.2021.03.049
7. Méni C, Bruneau J, Georgin-Lavialle S, Saché de Peufeilhoux LL, Damaj G, Hadj-Rabia S, et al. Paediatric mastocytosis: a systematic review of 1747 cases. Br J Dermatol. (2015) 172(3):642–51. doi: 10.1111/bjd.13567
8. Hartmann K, Escribano L, Grattan C, Brockow K, Carter MC, Alvarez-Twose I, et al. Cutaneous manifestations in patients with mastocytosis: consensus report of the European competence network on mastocytosis; the American academy of allergy, asthma & immunology; and the European academy of allergology and clinical immunology. J Allergy Clin Immunol. (2016) 137(1):35–45. doi: 10.1016/j.jaci.2015.08.034
9. Nettleship E, Tay W. Rare forms of urticaria. Br Med J. (1869) 2(455):323–4. doi: 10.1136/bmj.2.455.323
10. Sangster A. An anomalous mottled rash, accompanied by pruritus, factitious urticaria and pigmentation, “urticaria pigmentosa(?).”. Trans Clin Soc London. (1878) 11:161–3.
11. Drabent P, Polivka L, Agopian J, Duong Van Huyen JP, Thiebaut PA, Dubreuli P, et al. Establishing diagnostic criteria for mastocytosis in skin biopsies. Histopathology. (2022) 80(3):501–14. doi: 10.1111/his.14573
12. Hermans MAW, Pasmans SGMA, Arends NJT, van den Bosch TPP, van Daele PLA, van Doorn MBA, et al. Histopathological characteristics are instrumental to distinguish monomorphic from polymorphic maculopapular cutaneous mastocytosis in children. Clin Exp Dermatol. (2022) 47(9):1694–702. doi: 10.1111/ced.15262
13. Hao M, Zang P, Miller M, Cutler L, Worswick S. Herpes associated erythema multiforme: a retrospective study. Am J Emerg Med. (2020) 38(12):2761.e1–e3. doi: 10.1016/j.ajem.2020.05.084
14. Alshiyab DM, Al-Qarqaz FA, Alhaje E, Mayou JA, Jaradat S, Asaad A, et al. Skin manifestations among patients admitted with COVID-19: a cross-sectional study at a university-based tertiary hospital in Jordan. Clin Cosmet Investig Dermatol. (2023) 16:1331–40. doi: 10.2147/CCID.S408958
15. Yousefian M, Khadivi A. Occurrence of erythema multiforme following COVID-19 vaccination: a review. Clin Exp Vaccine Res. (2023) 12(2):87–96. doi: 10.7774/cevr.2023.12.2.87
16. Etaee F, Eftekharian M, Naguib T, Daveluy S. Erythema multiforme in COVID-19 patients and following COVID-19 vaccination: manifestations, associations and outcomes. J Eur Acad Dermatol Venereol. (2022) 36(7):e524–30. doi: 10.1111/jdv.18063
17. Su JR, Haber P, Ng CS, Marquez PL, Dores GM, Perez-Vilar S. Erythema multiforme, Stevens Johnson syndrome, and toxic epidermal necrolysis reported after vaccination, 1999–2017. Vaccine. (2020) 38(7):1746–52. doi: 10.1016/j.vaccine.2019.12.028
18. Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practising dermatologist. Int J Dermatol. (2012) 51(8):889–902. doi: 10.1111/j.1365-4632.2011.05348.x
19. Hughey LC. Approach to the hospitalized patient with targetoid lesions. Dermatol Ther. (2011) 24(2):196–206. doi: 10.1111/j.1529-8019.2011.01395.x
20. Zoghaib S, Kechichian E, Souaid K, Soutou B, Helou J, Tomb R. Triggers, clinical manifestations, and management of pediatric erythema multiforme: a systematic review. J Am Acad Dermatol. (2019) 81(3):813–22. doi: 10.1016/j.jaad.2019.02.057
21. Huff JC, Weston WL, Tonnesen MG. Erythema multiforme: a critical review of characteristics, diagnostic criteria, and causes. J Am Acad Dermatol. (1983) 8(6):763–75. doi: 10.1016/S0190-9622(83)80003-6
22. Kolkhir P, Grakhova M, Bonnekoh H, Krause K, Maurer M. Treatment of urticarial vasculitis: a systematic review. J Allergy Clin Immunol. (2019) 143(2):458–66. doi: 10.1016/j.jaci.2018.09.007
23. Kassim JM, Igali L, Levell NJ. A 14-year paraneoplastic rash: urticarial vasculitis and dermal binding bullous pemphigoid secondary to chronic lymphocytic leukaemia. Clin Exp Dermatol. (2015) 40(4):391–4. doi: 10.1111/ced.12553
24. Shah D, Rowbottom AW, Thomas CL, Cumber P, Chowdhury MMU. Hypocomplementaemic urticarial vasculitis associated with non-Hodgkin lymphoma and treatment with intravenous immunologlobulin. Br J Dermatol. (2007) 157(2):392–3. doi: 10.1111/j.1365-2133.2007.07983.x
25. Carlson JA, Chen KR. Cutaneous vasculitis update: small vessel neutrophilic vasculitis syndromes. Am J Dermatopathol. (2006) 28(6):486–506. doi: 10.1097/01.dad.0000246646.45651.a2
26. Marzano AV, Maronese CA, Genovese G, Ferrucci S, Moltrasio C, Asero R, et al. Urticarial vasculitis: clinical and laboratory findings with a particular emphasis on differential diagnosis. J Allergy Clin Immunol. (2022) 149(4):1137–49. doi: 10.1016/j.jaci.2022.02.007
27. Giang J, Seelan MAJ, van Doorn MBA, Rissmann R, Prens EP, Damman J. Complement activation in inflammatory skin diseases. Front Immunol. (2018) 9:639. doi: 10.3389/fiimu.2018.00639
28. McDuffie FC, Sams WM Jr, Maldonado JE, Andreini PH, Conn DL, Samayoa EA. Hypocomplementaemia with cutaneous vasculitis and arthritis. Possible immune complex syndrome. Mayo Clin Proc. (1973) 48(5):340–8.4267356
29. Sanchez NP, Winkelmann RK, Schroeter AL, Dicken CH. The clinical and histopathologic spectrums of urticarial vasculitis: study of forty cases. J Am Acad Dermatol. (1982) 7(5):599–605. doi: 10.1016/S0190-9622(82)70139-2
30. Wisnieski JJ, Jones SM. Comparison of autoantibodies to the collagen-like region of C1q in hypocomplementemic urticarial vasculitis syndrome and systemic lupus erythematosus. J Immunol. (1992) 148(5):1396–403. doi: 10.4049/jimmunol.148.5.1396
31. Kallenberg CGM. Anti-C1q autoantibodies. Autoimmun Rev. (2008) 7(8):612–5. doi: 10.1016/j.autrev.2008.06.006
32. Potlukova E, Kralikova P. Complement component C1q and anti-C1q antibodies in theory and in clinical practice. Scand J Immunol. (2008) 67(5):423–30. doi: 10.1111/j.1365-3083.2008.02089.x
33. Bigler C, Schaller M, Perahud I, Osthoff M, Trendelenburg M. Autoantibodies against complement C1q specifically target C1q bound on early apoptotic cells. J Immunol. (2009) 183(5):3512–21. doi: 10.4049/jimmunol.0803573
34. David C, Jachiet M, Pineton de Cambrun M, Gamez AS, Mehdaoui A, Zenone T, et al. Chronic obstructive airway disease associated with hypocomplementemic urticarial vasculitis. J Allergy Clin Immunol Pract. (2020) 8(9):3222–4. doi: 10.1016/j.jaip.2020.05.031
35. Mehregan DR, Hall MJ, Gibson LE. Urticarial vasculitis: a histopathologic and clinical review of 72 cases. J Am Acad Dermatol. (1992) 26(3):441–8. doi: 10.1016/0190-9622(92)70069-R
36. Ion O, Obrisca B, Ismail G, Sorohan B, Balanica S, Mircescu G, et al. Kidney involvement in hypocomplementemic urticarial vasculitis syndrome – a case-based review. J Clin Med. (2020) 9(7):2131. doi: 10.3390/jcm9072131
37. Messiaen T, van Damme B, Kuypers D, Maes B, Vanrenterghem Y. Crescentic glomerulonephritis complicating the course of hypocomplementemic urticarial vasculitis. Clin Nephrol. (2000) 54(5):409–12.11105804
38. Puhl V, Bonnekoh H, Scheffel J, Hawro T, Weller K, von den Driesch P, et al. A novel histopathological scoring system to distinguish urticarial vasculitis from chronic spontaneous urticaria. Clin Transl Allergy. (2021) 11(2):e12031. doi: 10.1002/clt2.12031
39. Rasmussen ER, Valente de Freitas P, Bygum A. Urticaria and prodromal symptoms including erythema marginatum in Danish patients with hereditary angioedema. Acta Derm Venereol. (2016) 96(3):373–6. doi: 10.2340/00015555-2233
40. Magerl M, Doumoulakis G, Kalkounou I, Weller K, Church MK, Kreuz W, et al. Characterisation of prodromal symptoms in a large population of patients with hereditary angioedema. Clin Exp Dermatol. (2014) 39(3):298–303. doi: 10.1111/ced.12285
41. Ohsawa I, Fukunage A, Imamura S, Iwamoto K, Tanaka A, Hide M, et al. Survey of actual conditions of erythema marginatum as a prodromal symptom in Japanese patients with hereditary angioedema. World Allergy Organ J. (2021) 14(12):100511. doi: 10.1016/j.waojou.2021.100511
42. Rosen FS, Pensky J, Donaldson V, Charache P. Hereditary angioneurotic edema: two genetic variants. Science. (1965) 148(3672):957–8. doi: 10.1126/science.148.3672.957
43. Bork K, Barnstedt SE, Koch P, Traupe H. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet. (2000) 356(9225):213–7. doi: 10.1016/S0140-6736(00)02483-1
44. Bork K, Wulff K, Steinmüller-Magin L, Braenne I, Staubach-Renz P, Witzke G, et al. Hereditary angioedema with a mutation in the plasminogen gene. Allergy. (2018) 73(2):442–50. doi: 10.1111/all.13270
45. Bafunno V, Firinu D, D’Apolito M, Cordisco G, Joffredo S, Leccese A, et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol. (2018) 141(3):1009–17. doi: 10.1016/j.jaci.2017.05.020
46. Bork K, Wulff K, Rossmann H, Steinmüller-Magin L, Braenne I, Witzke G, et al. Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the N-terminal cleavage site of bradykinin. Allergy. (2019) 74(12):2479–81. doi: 10.1111/all.13869
47. Ariano A, D’Apolito M, Bova M, Bellanti F, Loffredo S, D’Andrea G, et al. A myoferlin gain-of-function variant associates with a new type of hereditary angioedema. Allergy. (2020) 75(11):2989–92. doi: 10.1111/all.14454
48. Bork K, Wulff K, Möhl BS, Steinmüller-Magin L, Witzke G, Hardt J, et al. Novel hereditary angioedema linked with a heparan sulfate 3-O-sulfotransferase 6 gene mutation. J Allergy Clin Immunol. (2021) 148(4):1041–8. doi: 10.1016/j.jaci.2021.01.011
49. Kossard S, Hamann I, Wilkinson B. Defining urticarial dermatitis: a subset of dermal hypersensitivity reaction pattern. Arch Dermatol. (2006) 142(1):29–34. doi: 10.1001/archderm.142.1.29
50. Fung MA. The clinical and histopathologic spectrum of “dermal hypersensitivity reactions”, a nonspecific histologic diagnosis that is not very useful in clinical practice, and the concept of a “dermal hypersensitivity reaction pattern”. J Am Acad Dermatol. (2002) 47(6):898–907. doi: 10.1067/mjd.2002.120908
51. Bernard P, Antonicelli F. Bullous pemphigoid: a review of its diagnosis, associations and treatment. Am J Clin Dermatol. (2017) 18(4):513. doi: 10.1007/s40257-017-0264-2
52. Schmidt E, della Torre R, Borradori L. Clinical features and practical diagnosis of bullous pemphigoid. Dermatol Clin. (2011) 29(3):427–38. doi: 10.1016/j.det.2011.03.010
53. Kridin K, Bergman R. Assessment of the prevalence of mucosal involvement in bullous pemphigoid. JAMA Dermatol. (2019) 155(2):166–71. doi: 10.1001/jamadermatol.2018.5049
54. Lamberts A, Meijer JM, Pas HH, Diercks GFH, Horváth B, Jonkman MF. Nonbullous pemphigoid: insights in clinical and diagnostic findings, treatment responses, and progress. J Am Acad Dermatol. (2019) 81(2):355–63. doi: 10.1016/j.jaad.2019.04.029
55. Guerrois F, Hassan E, Bettuzzi T, Seta V, Goulvestre C, Jelti L, et al. Bullous pemphigoid: three main clusters defining 3 outcome profiles. J Am Acad Dermatol. (2022) 87(2):359–65. doi: 10.1016/j.jaad.2022.04.029
56. Shih YC, Wang B, Yuan H, Zheng J, Pan M. Role of BP230 autoantibodies in bullous pemphigoid. J Dermatol. (2020) 47(4):317–26. doi: 10.1111/1346-8138.15251
57. Ishiura N, Fujimoto M, Watanabe R, Nakashima H, Kuwano Y, Yazawa N, et al. Serum levels of IgE anti-BP180 and anti-BP230 autoantibodies in patients with bullous pemphigoid. J Dermatol Sci. (2008) 49(2):153–61. doi: 10.1016/j.jdermsci.2007.08.008
Keywords: urticaria, mimickers of urticaria, cutaneous mastocytosis, erythema multiforme, urticaria vasculitis, erythema marginatum, urticarial dermatitis, bullous pemphigoid
Citation: Fok JS and Katelaris CH (2023) Urticaria and mimickers of urticaria. Front. Allergy 4:1274031. doi: 10.3389/falgy.2023.1274031
Received: 7 August 2023; Accepted: 5 September 2023;
Published: 28 September 2023.
Edited by:
Murat Türk, Kayseri City Education and Research Hospital, TürkiyeReviewed by:
Andaç Salman, Acibadem University, Türkiye© 2023 Fok and Katelaris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Shen Fok jieshen.fok@easternhealth.org.au
†These authors have contributed equally to this work