 Dominique Missiakas
Dominique Missiakas Volker Winstel
Volker Winstel- 1Howard Taylor Ricketts Laboratory, Department of Microbiology, University of Chicago, Lemont, IL, United States
- 2Research Group Pathogenesis of Bacterial Infections, TWINCORE, Centre for Experimental and Clinical Infection Research, a joint venture between the Hannover Medical School and the Helmholtz Centre for Infection Research, Hannover, Germany
- 3Institute of Medical Microbiology and Hospital Epidemiology, Hannover Medical School, Hannover, Germany
Host cell death programs are fundamental processes that shape cellular homeostasis, embryonic development, and tissue regeneration. Death signaling and downstream host cell responses are not only critical to guide mammalian development, they often act as terminal responses to invading pathogens. Here, we briefly review and contrast how invading pathogens and specifically Staphylococcus aureus manipulate apoptotic, necroptotic, and pyroptotic cell death modes to establish infection. Rather than invading host cells, S. aureus subverts these cells to produce diffusible molecules that cause death of neighboring hematopoietic cells and thus shapes an immune environment conducive to persistence. The exploitation of cell death pathways by S. aureus is yet another virulence strategy that must be juxtaposed to mechanisms of immune evasion, autophagy escape, and tolerance to intracellular killing, and brings us closer to the true portrait of this pathogen for the design of effective therapeutics and intervention strategies.
Introduction
Human innate immune defenses substantially contribute to microbial clearance during infection (1, 2). Primary defenses encompass mechanisms that include the biosynthesis of antimicrobial peptides on the skin or mucosal surfaces, the recruitment of immune cells to infectious foci, and the activation of the complement system and coagulation cascade (2–4). Programmed cell death modalities represent additional key mechanisms that affect host-microbe interaction and infection control (5). Amongst cell death programs, conventional apoptosis, regulated necrosis (necroptosis), and pyroptosis have thoroughly been described to reveal unique signaling routes for initiation and execution of cell death (6, 7). Signaling and ensuing death modes are governed by the nature of the infection and the pathogenic attributes of the invading microbe. For example, apoptosis is often activated to release intracellular pathogens from infected host cells or tissues (8). In this manner, the host removes a preferred niche for initial replication, and simultaneously exposes the pathogen to extracellular immune cell defenses without causing inflammation. On the contrary, necroptosis and pyroptosis are highly inflammatory and impact immune cell trafficking as well as clinical syndromes and host-mediated clearance of pathogenic microorganisms (5).
Many human pathogens have evolved sophisticated strategies to modulate or subvert host cell death programs during infection (9). Specifically, microbes that infiltrate host cells and replicate intracellularly suppress death signaling pathways to escape extracellular immuno-surveillance (10, 11). In this manner, intracellular bacterial pathogens such as Mycobacterium tuberculosis or Legionella pneumophila maintain their proliferative niche to cause persistent infections (12–14). Yet, not all pathogens block cell death modalities upon host invasion. Some infectious agents, such as Staphylococcus aureus, induce or exploit programmed cell death to establish infection and disseminate in the host. S. aureus is the most frequently encountered agent of superficial skin and soft tissue infections and occasionally causes invasive diseases in humans. Once disseminated through blood stream infection, S. aureus is able to establish replication foci in almost any organ (Figure 1) (15, 16). S. aureus deploys an arsenal of virulence factors with potent immunomodulatory or toxigenic properties that modulate programmed cell death in professional and non-professional phagocytes thereby affecting clinical syndromes and various diseases in human or animal hosts (Figure 1) (17, 18). Overall, this remarkable microbe has evolved to manipulate all known principal mechanisms of programmed cell death, including apoptosis and pro-inflammatory necroptotic or pyroptotic cell death.
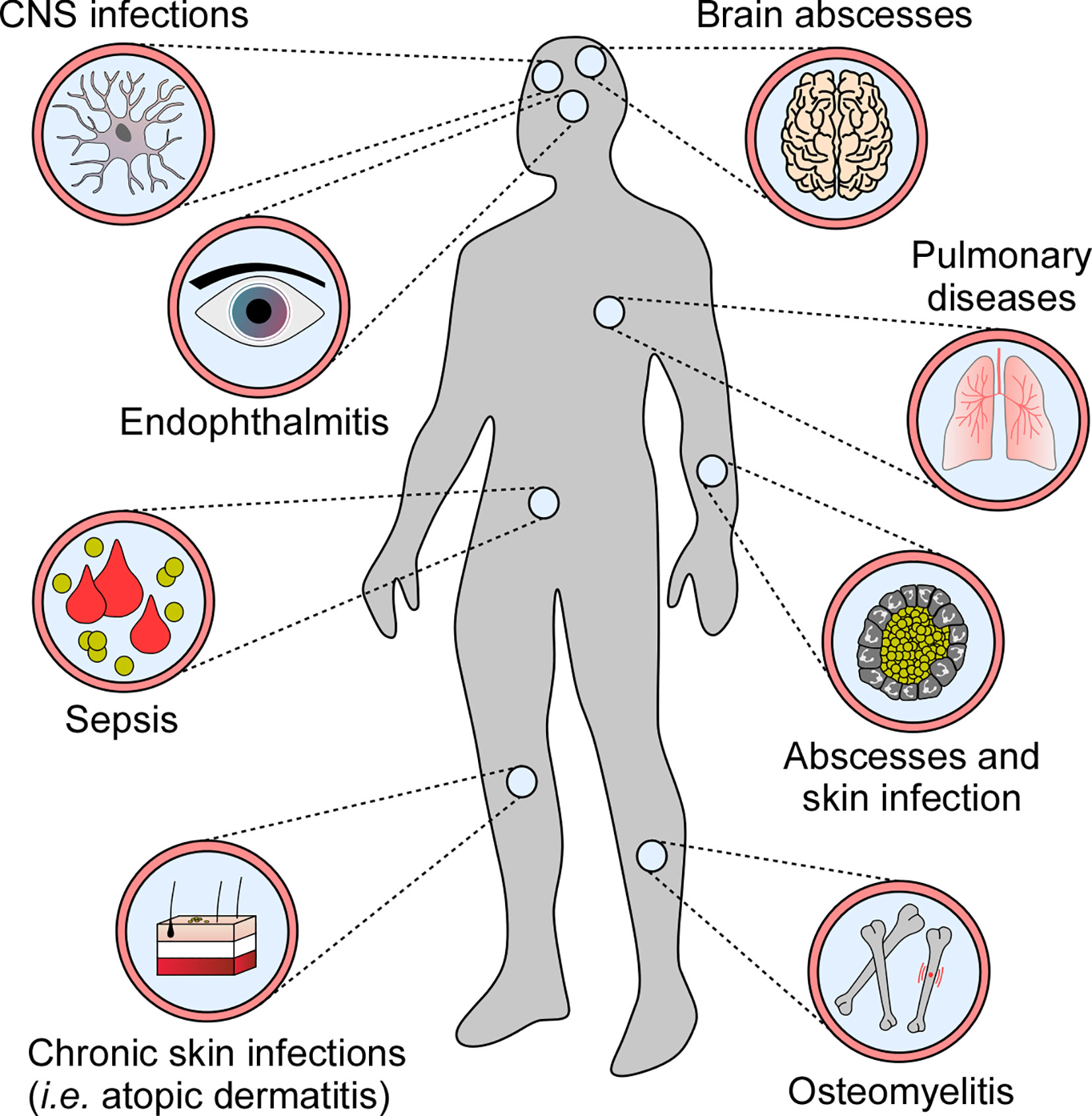
Figure 1 Staphylococcal diseases associated with programmed cell death. S. aureus exploits programmed cell death to cause various diseases in human and animal hosts.
Herein, we summarize various cell death modalities and their impact on the pathogenesis of S. aureus infections. We provide an overview of S. aureus-derived products that promote or avert programmed cell death signaling in host cells. Finally, we highlight staphylococcal tactics for the manipulation of autophagy, a cell death-associated cytoplasmic degradation mechanism that sustains cellular homeostasis and survival.
Apoptosis and Apoptotic Signaling Pathways
Apoptosis is an essential mechanism attributed to various physiological events. Apoptosis is considered an important component of multiple cellular processes and plays a significant role during normal development, organ shaping, homeostasis, and aging (19). Apoptosis is also favored by stress, lack of nutrition, and several other pathological conditions (19). Earlier work identified key genetic elements and two major signaling routes that regulate apoptosis in mammalian cells: the intrinsic (mitochondrial) and extrinsic (death receptor-mediated) pathways of apoptosis (Figure 2) (19). The extrinsic pathway is triggered by external signals and transmembrane death receptors (i.e., FasR or TNFR1) for activation of the death-inducing signaling complex (DISC) and initiator caspases-8 and -10; the intrinsic pathway is induced by internal stimuli, subcellular stress, and the release of apoptogenic proteins from injured mitochondria (Figure 2) (19). Microbial infections, DNA damage, cytotoxic stimuli, and various other pro-apoptotic signaling molecules promote permeabilization of the mitochondrial outer membrane, in a process mainly controlled by proteins of the Bcl-2 family (i.e., Bcl-2-associated X protein (Bax)) (20–22). Cellular stress favors oligomerization of Bax and Bak (Bcl-2-antagonist killer 1) and subsequent formation of pores in mitochondrial membranes (21, 23). Perforated mitochondria release cytochrome c and other pro-apoptotic proteins into the cytosolic space (19, 21, 24). Voltage-dependent anion-selective channels (VDAC) may enhance the release of mitochondrial pro-apoptotic factors by interacting with dedicated Bcl-2 family proteins (25–27). Cytosolic cytochrome c, together with dATP and the apoptotic protease activating factor 1 (APAF1), trigger the formation of the ultra-large apoptosome complex that activates the initiator caspase-9 (28, 29). Once caspase-9 (or caspases-8 or -10 in case of the extrinsic pathway of apoptosis) is activated, effectors caspases-3, -6, and -7 are proteolytically processed and converted to mature proteins that degrade defined target substrates; the ultimate result culminates with cell death exhibiting typical morphological features of apoptosis: membrane blebbing, cell shrinkage, DNA fragmentation, nuclear condensation, and formation of apoptotic bodies (Figure 2) (19).
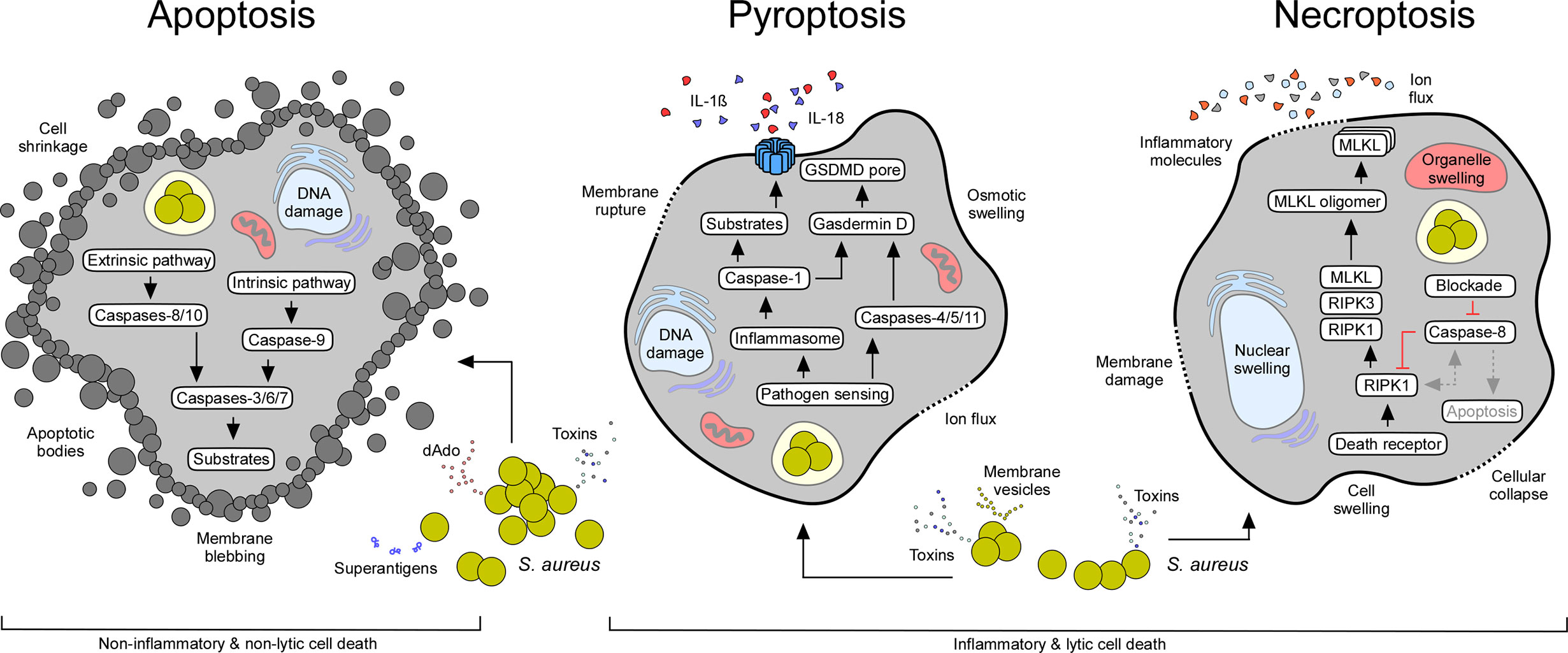
Figure 2 Staphylococcal interference with host cell death machineries. All major cell death modes including apoptosis, pyroptosis, and necroptosis may occur in response to extra- or intracellular staphylococci and their exoproducts (see Table 1). While apoptotic cell death is immunologically silent, pyroptosis and necroptosis cause strong inflammatory responses due to the release of pro-inflammatory molecules from injured host cells. Characteristic features and canonical signaling pathways of cell death modalities are indicated.
Apoptotic Cell Death in Response to Staphylococcus aureus Infections
Apoptotic cell death of hematopoietic and non-hematopoietic cells plays a significant role during S. aureus disease pathogenesis. During infection, S. aureus provokes apoptosis in a broad spectrum of target cells as a means to invade tissues, and to antagonize host immune defenses (18, 30). Depending on the type of tissue and staphylococcal isolate, apoptosis may occur via extrinsic-, intrinsic-, or caspase-2-mediated apoptotic signaling (31–36). S. aureus produces a vast array of pro-apoptotic virulence factors that predominantly encompass potent toxins and superantigens (Table 1) (17). Genetic variability amongst S. aureus isolates increases the repertoire of toxins and superantigens. All of these factors are secreted into the extracellular milieu and are endowed with membrane-damaging or toxigenic properties that interfere with apoptotic signaling cascades (17, 76). For example, the staphylococcal pore-forming toxins α-toxin, leukocidin AB (LukAB), or the Panton-Valentine-leukocidin (PVL), have been shown to prime apoptotic cell death in professional phagocytes and other cells (31–33, 36, 48, 49). Pore-forming toxin-mediated apoptosis involves potassium efflux from damaged cells and caspase-2-initiated cell death, or breakdown of the mitochondrial membrane potential, ultimately leading to the release of apoptogenic factors (e.g., cytochrome c) and activation of intrinsic death signaling pathway (31–33, 36). Staphylococcal superantigens (i.e., enterotoxin B) interact with T-cell receptors via major histocompatibility complex (MHC-II) molecules to stimulate biosynthesis and release of apoptogenic factors such as TNF-α, FasL, or IFN-γ (35, 44, 55). In this fashion, S. aureus triggers a pro-apoptotic milieu that induces extrinsic apoptosis in adjacent host target cells. Overall, toxin-mediated activation of apoptosis and subsequent killing of phagocytes eliminates primary host defenses essential for pathogen clearance. Pore-forming toxins are also thought to be key for the successful facultative intracellular lifestyle of S. aureus in non-professional phagocytes. Specifically, internalization of staphylococci by epithelial cells, endothelial cells, fibroblasts, keratinocytes, or osteoblasts can stimulate apoptotic cell death signaling (Figure 2) (40, 50, 77–82). In this manner, S. aureus not only escapes from host immune cell responses but also promotes tissue injury, and subsequent infiltration into deeper tissues, organs, or circulating body fluids. Following blood stream dissemination, S. aureus can successfully invade organ tissues to seed abscess lesions by initiating apoptotic death of surrounding cells in a manner independent of pore-forming toxins or superantigens (Table 1). S. aureus abscess formation involves two secreted enzymes, staphylococcal nuclease and adenosine synthase A (AdsA), that together convert neutrophil extracellular traps (NETs) into deoxadenosine (dAdo), a pro-apoptotic molecule, which kills phagocytes (37). dAdo-intoxication of macrophages involves uptake of dAdo by the human equilibrative nucleoside transporter 1 (hENT1), subsequent targeting of the mammalian purine salvage pathway, and signaling via dATP formation to activate caspase-3-dependent apoptosis and immune cell death (37–39). In this manner, macrophages are excluded from abscess lesions without causing inflammation thereby promoting the establishment of invasive disease (37, 39). More recently, Stelzner and colleagues discovered that intracellular S. aureus elaborates Staphopain A, a secreted cysteine protease, to trigger apoptosis in epithelial cells after translocation to the host cell cytosol (53). Staphopain B and the type-VII secretion system effector EsxA may also interfere with apoptotic cell death of human cells (Table 1) (47, 51, 52, 54). In summary, S. aureus exploits apoptosis to incapacitate macrophages and other host cells without provoking inflammatory responses; this facilitates infiltration of the bacteria in tissues and the establishment of persistent lesions filled with replicating staphylococci.
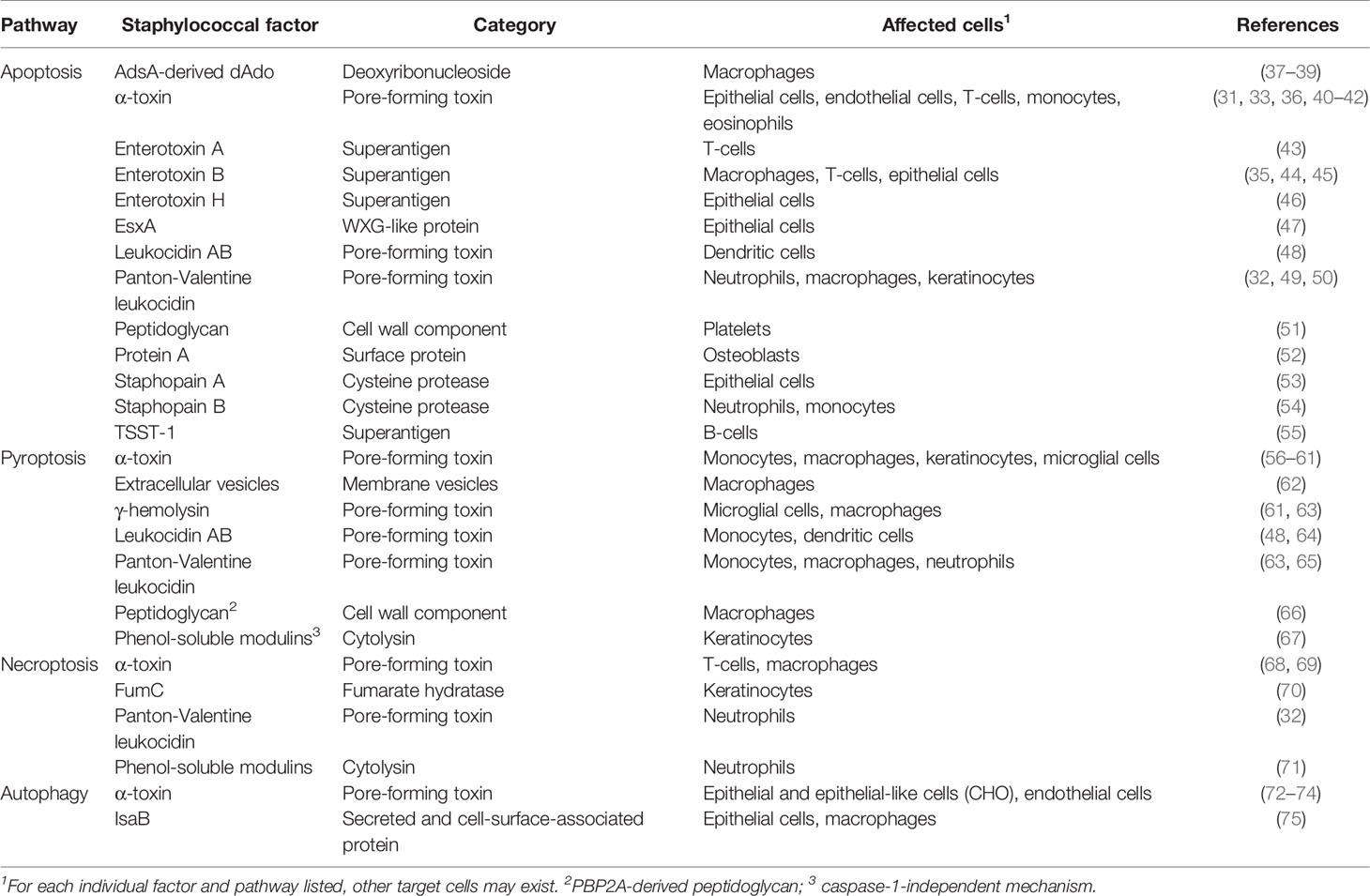
Table 1 Selected staphylococcal factors interfering with programmed cell death and autophagic signaling pathways.
Pyroptosis and the Inflammasome
Unlike apoptosis, pyroptosis denotes a highly inflammatory state that largely depends on the activation of interleukin-1β (IL-1β)-converting enzyme also known as caspase-1 (Figure 2) (6). Caspase-1 is synthesized as an inactive zymogen in mammalian cells and was the first cysteine-dependent aspartate-specific protease (caspase) discovered in scientific history (83). Processing and subsequent proteolytic cleavage of caspase-1 occurs within the inflammasome, a supramolecular complex that encompasses a member of NOD-like receptors (NLRs) (Figure 2) (6, 84). NLRs contain carboxy-terminal leucine rich repeats (LRR), a structural feature shared with Toll-like receptors (TLRs), which evolved to sense a large set of pathogen-associated danger signals, including bacterial or viral nucleic acids (85–88). In addition, inflammasome-associated NLRs are endowed with a variable N-terminal region that consists of a caspase activation and recruitment domain (CARD), or a pyrin (PYD) subunit that interacts with a CARD-domain containing adaptor protein (ASC) (CARD domains facilitate binding and proteolytic cleavage of caspase-1) (88–92). Following processing, catalytically active caspase-1 cleaves pro-forms of IL-1β, IL-18, and IL-33 into biologically active and secreted cytokines, ultimately leading to strong pro-inflammatory responses that dictate the recruitment of immune cells and pathophysiological outcome of disease (Figure 2) (6). Inflammasome activated caspase-1 also cleaves pro-Gasdermin D, the actual executor of pyroptosis (93, 94). Processing of Gasdermin D leads to the release of a plasma membrane pore-forming subunit (GSDMD-N domain) that interacts with acidic phospholipids found on the inner leaflet of mammalian plasma membranes (Figure 2) (95, 96). Together with distinct mechanisms such as microvesicle shedding (97), GSDMD-N-derived plasma membrane pores facilitate the rapid release of the aforementioned pro-inflammatory cytokines and intracellular molecules into the extracellular milieu, and ultimately drive swelling and osmotic lysis of host cells (Figure 2) (95, 96). Other caspases may also trigger pyroptosis (98–100). For example, caspases-4, -5, -11, as well as apoptosis executor caspase-3, can process pro-Gasdermins directly upon stimulation, thus impacting pyroptotic cell death and its characteristic morphological features (94, 98–100).
S. aureus-Mediated Activation of Distinct Host Inflammasomes
S. aureus pathogenesis involves activation of distinct inflammasomes, a process that primarily depends on the infection site and staphylococcal stimulus involved. Pioneering work by Mariathasan et al. uncovered that exposure of NLRP3-deficient bone marrow-derived macrophages to replicating S. aureus drastically reduced the detectable amount of mature caspase-1, and secreted cytokines IL-1β and IL-18 (86). Subsequently, multiple other studies revealed that S. aureus pore-forming toxins contribute to this phenomenon, and trigger the formation of the NLRP3 inflammasome, cytokine release, pyroptosis, or pyroptotic-like cell death (Table 1). Purified α-toxin or α-toxin-containing S. aureus culture supernatants rapidly activate caspase-1 and NLRP3-dependent signaling in THP-1 cells or mouse macrophages (56, 57). Similarly, staphylococcal bi-component toxins LukAB or PVL induce processing of caspase-1 and release of pro-inflammatory cytokines by human phagocytes (64, 65). However, attempts to block the cognate host proteins with small molecule inhibitors only marginally suppresses α-toxin-, PVL-, or LukAB-mediated cell death (56, 64, 65). Even the genetic ablation of CASP1 cannot prevent bacterial pore-forming toxin-dependent killing of host cells, demonstrating that distinct mechanisms or cross talk between different cell death modalities may contribute to toxin-induced cell death (56, 64). In agreement with this notion, a drop in intracellular potassium as a result of K+ efflux caused by pore-forming toxins or activation of the lysosomal cysteine protease cathepsin B provoke assembly of the NLRP3 machinery and cytokine release (64, 65, 101, 102). More recent work revealed that pyroptotic cell death is also driven by S. aureus-derived membrane vesicles (MVs) (Table 1) (62). MVs deliver lipoproteins and pore-forming toxins along with other pro-pyroptotic effector molecules to host cells thereby stimulating TLR2-mediated priming of the NLRP3 inflammasome, ultimately leading to gasdermin D-dependent release of pro-inflammatory cytokines and pyroptotic cell death (62). Combined with the canonical secretory pathway, this dual strategy of toxin-mediated destruction of innate immune cells secures S. aureus survival in hosts and establishment of invasive disease.
S. aureus can also target the NLRP3 inflammasome and pyroptotic signaling in a subset of non-immune host cells. Keratinocytes, when exposed to live S. aureus, culture supernatants, or staphylococcal toxins, produce elevated levels of IL-1β and IL-18, and exhibit pyroptotic characteristics (58, 67). S. aureus-induced skin inflammation and severity of dermal disease has been correlated with stimulation of the inflammasome and cytokine signaling (57, 58, 67, 103). Neither wild-type mice infected subcutaneously with a panel of toxin-deficient S. aureus mutants, nor NLRP3-, ASC-, or CASP1-deficient animals infected with wild-type S. aureus elicit NLPR3-dependent inflammatory responses and cytokine signaling (57). As a result, ASC-/- or IL-1β-/- mice fail to recruit neutrophils and other phagocytes to infectious foci, and develop significantly enlarged lesions in an experimental model of S. aureus skin infection (Table 2) (103). In line with these observations, impaired expression of NLRP3, ASC, and CASP1 dampens neutrophil attraction in atopic dermatitis patients thereby increasing the risk of pathogen colonization and chronic skin inflammation (117). Yet, pyroptosis may also correlate with enhanced staphylococcal diseases, including traumatic osteomyelitis, central nervous system infections, and acute pneumonia (Figure 1) (59, 106, 118). As with skin infections, staphylococcal pulmonary disease and superinfections of lungs are associated with altered activity of the NLRP3 inflammasome (60, 107, 110). S. aureus-driven pneumonia induces additional inflammasome machineries such as NLRP6 (59). Of note, activation of the NLRP6 inflammasome during acute pneumonia negatively regulates pulmonary defenses, as NLRP6-/- mice accelerate neutrophil recruitment and display increased resistance to staphylococcal lung infection (Table 2) (59). Lastly, the NLRP7 inflammasome senses intracellular staphylococci and acetylated lipoproteins, restricting bacterial replication and dissemination of disease (119). However, the exact role of NLRP7 for S. aureus pathophysiology remains enigmatic. Collectively, S. aureus hijacks distinct inflammasomes and pyroptotic cell death modalities during infection, presumably to promote host invasion and immune evasion. Since S. aureus-induced activation of pyroptotic cell death elicits robust inflammatory and immune responses, pyroptosis may also contribute to host-mediated clearance of staphylococci.
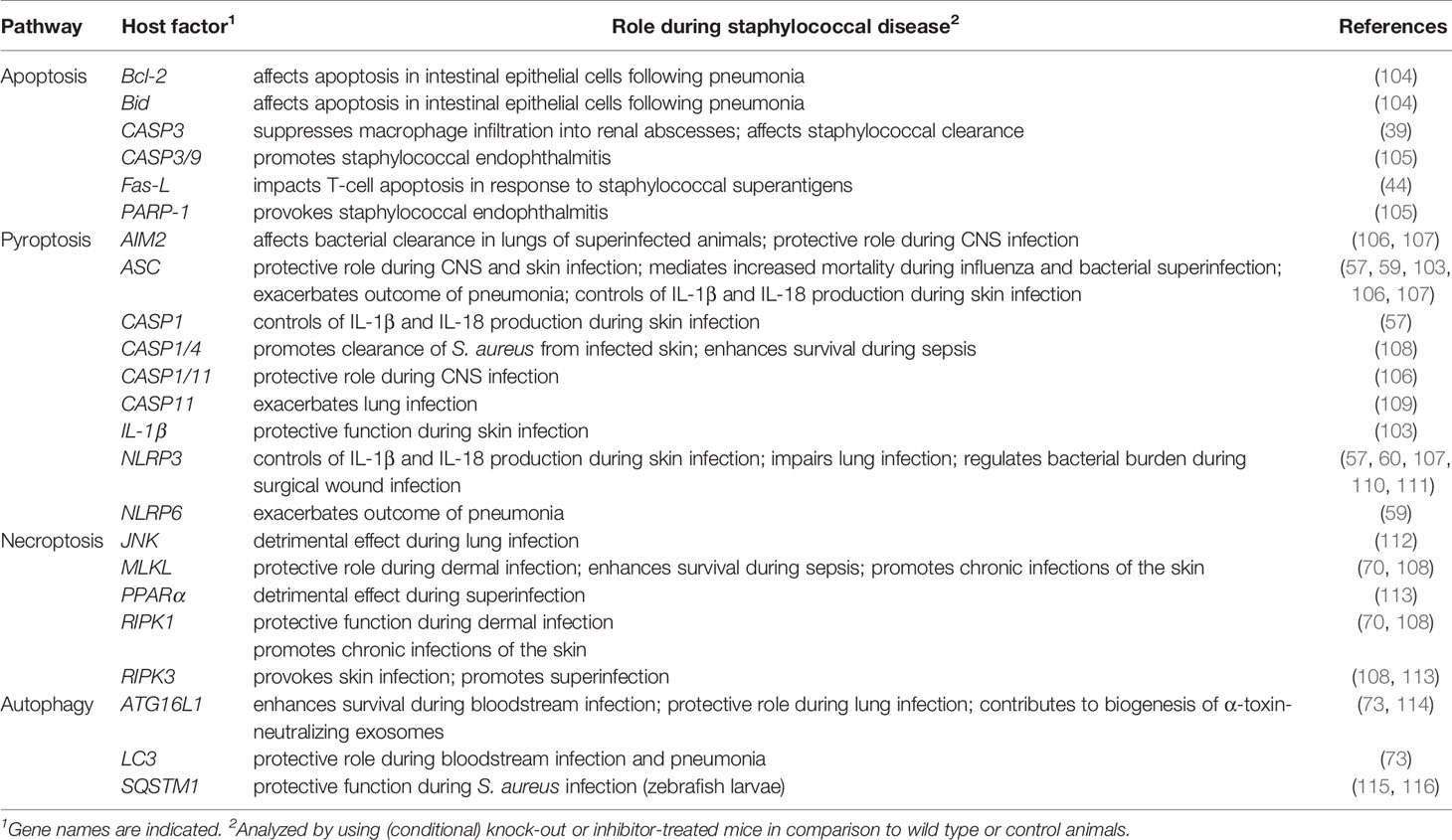
Table 2 Selected cell death- and autophagy-associated host genetic determinants affecting S. aureus pathogenesis in vivo.
Necroptotic Cell Death and Its Pathological Features
Necrosis stems from the Greek word “nekros” (dead body) and represents a passive and uncontrolled form of cell death. While initially considered to represent an accidental form of cell death that lacks a defined signaling network, recent work uncovered the existence of multiple pathways contributing to the control of necrosis (120). The prototypical form of regulated necrosis, necroptosis, requires several kinases, including the mixed lineage kinase domain-like protein (MLKL) and receptor-interacting protein kinases 1 and 3 (RIPK1, RIPK3); regulated necrosis also requires dedicated plasma membrane receptors and their ligands (Figure 2) (121–124). More precisely, necroptotic signaling largely depends on death receptor mediated signaling molecules (i.e., Fas or TNF) that interfere with their cognate plasma membrane receptors, leading to the formation of a stable, but short-lived RIPK1- and TRADD (TNFR1-associated death domain)-dependent receptor-bound complex I (122, 125–127). In addition to RIPK1 and TRADD, this multimeric complex encompasses cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1/cIAP2), TNF receptor-associated factor 2 (TRAF2) and TRAF5. Together, TRAF2 and TRAF5 mediate polyubiquitination of RIPK1 (126, 128–130). Ubiquitination of RIPK1 features the assembly of the inhibitor of nuclear factor-κB (NF-κB) kinase (IKK) complex, which promotes the upregulation of NF-κB pathway and several anti-apoptotic genes, including the FLICE-like inhibitory protein (FLIP) (120). However, deubiquitination of RIPK1 via cylindromatosis (CYLD) and other deubiquitinases destabilizes complex I, a crucial step that promotes interaction of RIPK1 with FADD (FAS-associated death domain), TRADD, RIPK3, pro-caspase-8, and the long isoform of FLIP (FLIPL) to form the TRADD-dependent complex II (126, 131–134). Subsequently, pro-caspase-8 and FLIPL form a heterodimer complex that cleaves and inactivates RIPK1, RIPK3, and CYLD to prevent necroptosis (135–139). Pro-caspase-8 homodimerization induces auto-proteolysis and formation of active caspase-8 that processes the apoptosis-executing caspases 3 and 7, ultimately promoting apoptotic cell death (120). Nevertheless, chemical or pathogen-induced blockade of caspase-8 provokes the complexation and autophosphorylation of RIPK1 and RIPK3 that leads to the assembly of an intracellular machinery designated necrosome (Figure 2) (140). Upon necrosome formation, downstream signaling leads to the recruitment of MLKL, a pseudokinase that interacts with the inner leaflet of plasma membranes in its phosphorylated state (123, 141–143). In this manner, MLKL disrupts the integrity of the cell thereby promoting necroptosis (Figure 2). Apart from death-receptor-mediated necroptosis, regulated necrosis can further be triggered by TLR-mediated signaling or certain intracellular stimuli that lead to the formation of non-classical necrosomes (144, 145). Moreover, DNA damage can activate RIPK3 and biogenesis of another necroptosis-executing multiprotein complex termed ripoptosome (146). Overall, necroptosis constitutes a caspase-independent form of programmed cell death that is morphologically characterized by massive organelle and cellular swelling, and rupture of plasma membranes (Figure 2). Hence, regulated necrosis causes robust inflammatory responses and severe tissue injury, thus affecting the pathophysiology of many infectious and non-infectious diseases.
Exploitation of Necroptotic Signaling by Staphylococcus aureus
The discovery of necroptotic signaling cascades enabled the staphylococcal research community to uncover the significance of necroptosis-dependent cell death in the pathophysiology of S. aureus diseases. Initial work aimed to identify microbial and host determinants that modulate necroptotic cell death during acute and persistent infections, specifically in the context of staphylococcal pulmonary disease (Tables 1 and 2). As expected, staphylococcal pore-forming toxins including α-toxin promote tissue damage and necroptotic cell death in immune and epithelial cells during lung infection (68, 147). Moreover, S. aureus phenol-soluble modulins (PSM peptides) constitute potent catalysts of necroptosis as these cytolytic peptides activate necroptotic death of host phagocytes via induction of MLKL phosphorylation, ultimately leading to exacerbated outcomes of staphylococcal pulmonary infections (71). Although most of these toxins have distinct receptors, all variants exhibit potent immunomodulatory properties that together trigger assembly of the necrosome, and subsequent necroptotic cell death (68, 148, 149). However, some of these studies revealed that S. aureus toxin-mediated necroptosis may directly interfere with pyroptotic signaling pathways. For example, it was found that the pharmacological inhibition of MLKL dampens caspase-1 activation and pyroptotic signaling in host cells upon staphylococcal stimulation (68, 150). Thus, it is not surprising that mice lacking the NLRP6 inflammasome exhibit both, reduced pyroptotic and necroptotic signaling following pathogen challenge (59). Nevertheless, pharmacological and genetic perturbation of key modulators of necroptosis such as MLKL, RIPK1, or RIPK3 can clearly protect human and murine macrophages as well as neutrophils from CA-MRSA strain USA300 and its secreted toxins (68, 148–151). In line with these findings, RIPK3 knock-out mice display increased resistance during experimental S. aureus lung infection, an effect attributed to anti-inflammatory CD206+ and CD200R+ alveolar macrophages that accumulate in lungs and may accelerate the clearance of staphylococci (Table 2) (68). Also, in vivo blockade of c-Jun N-terminal kinases (JNK1 and JNK2), both of which are known to trigger TNF- and TLR-induced necroptotic cell death, or genetic ablation of the peroxisome proliferator-activated receptor α (PPARα), a ligand-activated transcription factor and suppressor of NF-κB activation, rescued mice from fatal staphylococcal lung disease, even under conditions that mimic bacterial superinfections (Table 2) (112, 113). Zhou et al. exploited RNAIII-inhibiting peptide as an anti-virulence therapeutic approach to prevent PSM- and necroptosis-dependent lung injury in mice nasally infected with CA-MRSA strain USA300 (71). RNAIII is the effector of accessory gene regulator (Agr) and RNA-III inhibiting peptide blocks S. aureus quorum sensing and biogenesis of PSMs toxins during acute lung infection (71). Together, this compelling work underscores the importance and clinical relevance of necroptosis during S. aureus pulmonary disease, and suggests that staphylococcal exoproducts may simultaneously trigger distinct and genetically conserved cell death programs in mammalian cells to establish infection (Figure 2).
S. aureus can also trigger necroptosis in the absence of pore-forming toxins (Table 1). Wild-type S. aureus or its hemB variant stimulate host cell glycolytic activity and formation of mitochondrial reactive oxygen species in skin cells in a manner that promotes necroptotic cell death without the contribution of bacterial toxigenic molecules (70). Mutations in the hemB gene and other metabolic genes arise spontaneously during infection and are identified on laboratory medium as small colony variants (SCVs). SCVs represent auxotrophic subpopulations, which while less virulent, are able to persist within host cells and are associated with chronic infection (152). Intracellular hemB variants induce the biogenesis of bacterial fumarate hydratase that degrades cellular fumarate, a known inhibitor of the glycolytic pathway of mammalian cells (70). In this manner, SCVs activate necroptosis to promote persistence in skin cells (70). Induction of necroptosis may also represent a selective response of host keratinocytes to eradicate the invading pathogen as mice lacking key elements of the necroptotic signaling pathway such as MLKL exhibit significantly enlarged wounds and higher bacterial loads during S. aureus experimental skin infection (Table 2) (108). Although these animals recruited more immune cells to the primary skin lesion and produced elevated levels of pro-inflammatory cytokines due to excessive activation of caspase-1, they failed to clear replicating staphylococci (108). Consistent with these findings, MLKL-proficient animals display enhanced survival rates over time in a S. aureus murine bacteremia model, further suggesting that induction of necroptosis may be beneficial for the host (108). Collectively, these observations suggest that necroptosis may restrict hyper-inflammatory immune responses during skin or blood stream infections thereby serving as a protective mechanism that promotes bacterial eradication from infected hosts (108). However, S. aureus may also exploit the necroptotic signaling pathway to combat resident and recruited innate immune cells during acute or chronic infections.
Autophagy and Autophagic Cell Death
Autophagy (from Greek, “self-eating”) constitutes a highly conserved process that controls cellular development, homeostasis and survival (153). This housekeeping mechanism represents a protective platform and cellular recycling system that overcomes several pathological states or harmful conditions (153, 154). Comprehensive work uncovered mechanistic details and the existence of at least three forms of autophagy (micro- or macroautophagy, and chaperon-mediated autophagy), all of which rely on lysosome-based degradation of unnecessary or detrimental molecules (154–156). Herein, we focus on macroautophagy, which resembles the canonical form of autophagy and main mechanism known to interfere with staphylococcal infections (Figure 3).
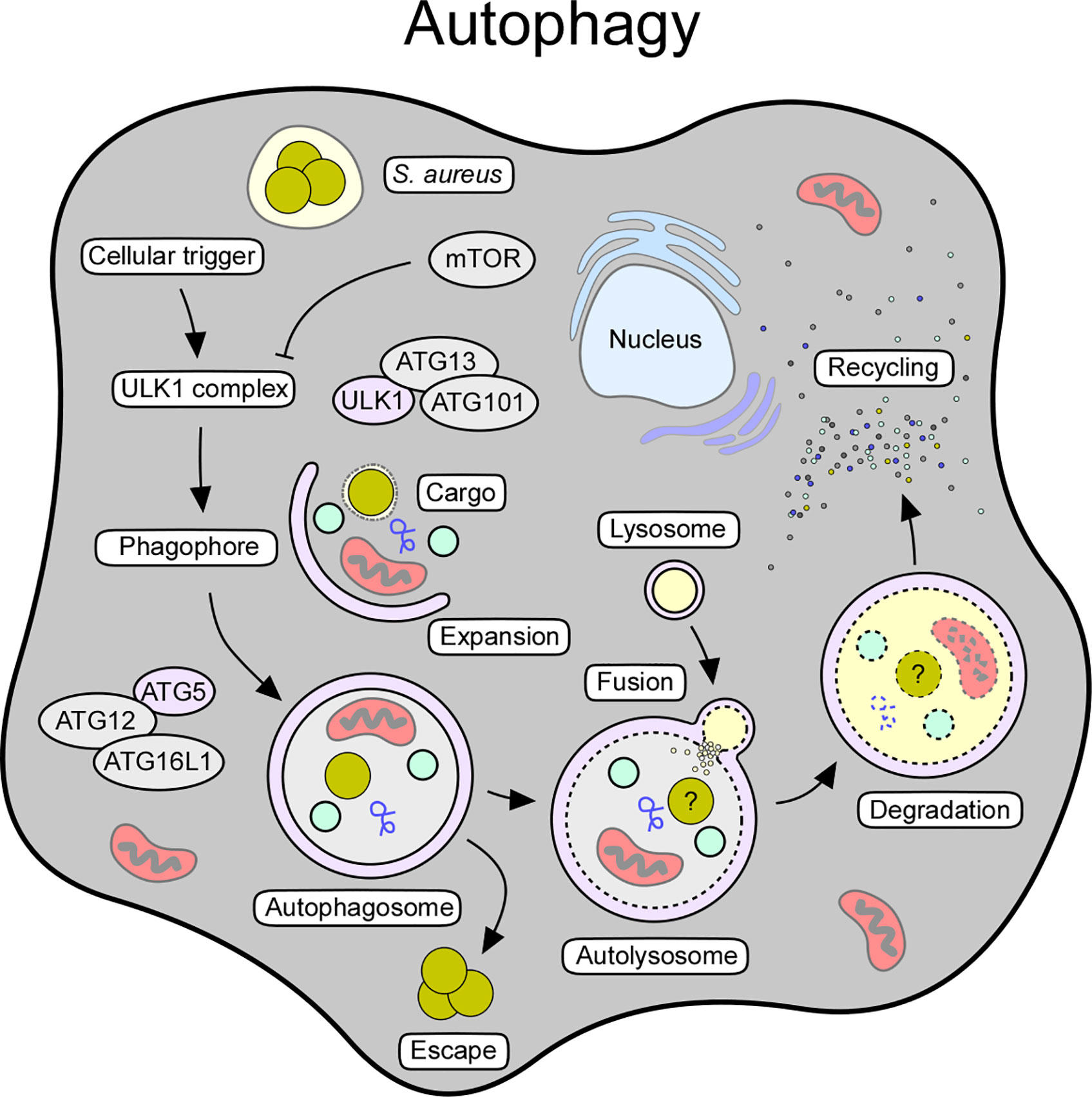
Figure 3 Cellular and molecular features of autophagy. Autophagy can be triggered by various stimuli, including starvation, oxidative stress, pathogens, or pathogen-derived products. The autophagic signaling process can also be initiated in response to intracellular staphylococci and their secreted toxins. Release of toxins and other virulence determinants presumably prevents maturation of autophagosomes thereby boosting staphylococcal escape from these structures. Key factors and crucial steps of the autophagy pathway are highlighted.
Macroautophagy (referred to as autophagy) requires a cellular trigger (i.e., starvation or oxidative stress) that abrogates mTOR-mediated suppression of autophagic signaling, thus leading to the assembly of the autophagy initiator complex (Unc-51-like kinase 1 [ULK1] complex) (153, 157) (Figure 3). This multimeric protein complex consists of ULK1, autophagy-related proteins (ATG)-13 and ATG101, and FIP200, a focal adhesion kinase family-interacting protein (153, 158). Following priming, the ULK1 complex phosphorylates AMBRA (activating molecule in Beclin-1-regulated autophagy protein 1) as part of the ATG14-, VPS15-, VPS34-, Beclin-1-, and p115-consisting PI3KC3 complex I (153, 159). Together with the ULK1 complex, this autophagy-modulating element initiates the biogenesis of phagophores, and subsequently promotes the generation of phosphatidylinositol-3-phosphate (PI3P), which serves as a docking scaffold for WD repeat domain phosphoinositide-interacting proteins (WIPIs) and other effector molecules (153, 154). Phagophore-associated WIPIs in turn recruit an array of ATG proteins (that is ATG16L1 and the ATG5-ATG12-ATG3 conjugate), which facilitate the ATG3-driven conjugation of ATG class 8 proteins such as LC3 (microtubule-associated protein light chain 3) to phosphatidylethanolamine (PE) (153, 160). In this manner, conjugated LC3 is lipidated and readily incorporated into autophagic membranes (153, 154). Membrane-associated LC3/ATG8 conjugates capture and recruit labeled (unwanted) molecules via selective autophagy receptors such as sequestosome 1 (SQSTM1/p62) (153, 154). Together with ATG9-positive vesicles and cellular membrane material, ATG8s further promote phagophore expansion and sealing around selected cargo, ultimately leading to the formation of the autophagosome (153, 154) (Figure 3). Once autophagosomes are fully assembled, ATG family proteins disassociate to enable maturation and fusion of the autophagosome with acidic hydrolases-containing lysosomes (153, 154) (Figure 3). In this manner, autolysosomes recycle cellular trash, intracellular pathogens, or damaged organelles into elementary building blocks required for macromolecule biosynthesis or energy supply (154). Thus, autophagy represents a major cytoprotective mechanism that sequesters cytoplasmic material in double-membraned vesicles for subsequent detoxification and degradation. Excessive induction of autophagy can also trigger autophagic cell death (ACD), as autophagy and other cell death signaling pathways are interconnected and together influence the fate of dying cells (161).
Staphylococcal Interference With Autophagic Signaling
Many intracellular pathogens such as Mycobacterium tuberculosis have developed refined strategies to antagonize autophagic signaling pathways during infection (162, 163). Owing to its ability to replicate in professional- and non-professional phagocytes, S. aureus is considered to be a facultative intracellular pathogen. This raises the possibility that S. aureus may suppress autophagy to cause persistent infection and chronic disease. Accumulating evidence suggests that S. aureus is indeed able to subvert autophagic responses (72, 164). Initial studies by Schnaith et al. demonstrated that S. aureus rapidly transits from endosophagosomes to LC3-positive autophagosomes upon invasion of HeLa cells (164). More recent work revealed that recruitment of S. aureus to phagophores requires SQSTM1/p62 and other autophagy receptor proteins that enable efficient tagging of staphylococci by neutrophils, fibroblasts, or keratinocytes (115, 116, 165). Although these investigations suggest that host cells may use autophagosomes to encircle intracellular staphylococci, S. aureus-containing autophagosomes cannot fuse with lysosomes and thereby fail to clear intracellular staphylococci (164). Autophagic vesicles and non-acidified phagosomes rather constitute a survival containment for host cell-engulfed staphylococci; S. aureus is able to exit these vesicles by secreting autophagosome-damaging toxins and other virulence determinants (116, 165, 166) (Figure 3). Indeed, α-toxin-proficient S. aureus or purified α-Hemolysin promote the initiation of an autophagic response but prevent maturation of autophagosomes (Table 1) (72). Concomitantly, autophagosome-associated staphylococci block autophagosome maturation by initiating the phosphorylation of the mitogen-activated protein kinase MAPK14 (p38α) (MAPK14/p38α) (165). Upon phosphorylation, activated MAPK14 traffics to autophagosomes where it inhibits autophagosome maturation and fusion with acidified lysosomes (167). S. aureus also deploys the immuno-dominant surface antigen B (IsaB), a secreted and cell surface-associated protein, to limit the autophagic flux in host cells thereby enhancing intra-host cell survival (Table 1) (75). Since isaB expression levels correlate with improved host colonization, IsaB-mediated subversion of autophagy may also promote host-to-host transmission of highly transmissible MRSA isolates (75). Exploitation of autophagic responses has further been observed in dendritic cells, where staphylococci accumulate in autophagosomes in an Agr-dependent manner as well as in keratinocytes or bovine mammary epithelial cells (168–170). S. aureus-mediated manipulation of the central carbon metabolism of host cells, as recently described for HeLa cells exposed to CA-MRSA strain USA300, promotes autophagic signaling and intracellular proliferation of bacteria (171). Specifically, NMR- and MS-based profiling of MRSA-infected cells revealed the conspicuous metabolic starvation of infected host cells, a typical trigger of autophagy sensed by the autophagy master regulator mTOR (171). Autophagy also contributes to innate immune cell defenses during staphylococcal disease pathogenesis, particularly in the context of infection control and tolerance to bacterial toxins (Table 2). Recent work by Gibson and colleagues suggested that autophagy governs cytosolic surveillance of replicating staphylococci in neutrophils (115). Using a zebra fish infection model, the investigators demonstrated that SQSTM1/p62 along with LC3 targets neutrophil-engulfed staphylococci for subsequent degradation in vivo, thus illustrating the protective potential of autophagy during staphylococcal infections (115). In agreement with this study, Maurer et al. discovered that autophagy diminishes host susceptibility to acute S. aureus infections, as autophagy-deficient mice (here: ATG16L1-hypomorph [ATG16L1HM] or LC3-/- mice) display hypersensitivity towards S. aureus (73). Remarkably, increased mortality of ATG16L1HM mice during both, sepsis or acute pneumonia, correlated with the biogenesis of staphylococcal α-toxin and its endothelial-damaging properties, and with elevated protein levels of ADAM10, the α-toxin receptor (73). Subsequent work by the same group uncovered that TLR9-sensed bacterial and CpG DNA along with ATG16L1 and other ATG proteins promote the release of ADAM10-containing exosomes during infection (114). These secreted exosomes capture and neutralize α-toxin and other bacterial toxins, a striking feature that protects the host from toxinosis and severe clinical syndromes (114). Together, these studies uncovered a crucial role of autophagy during staphylococcal infections. While S. aureus is able to hijack autophagosomes to elude from phagocytic killing and innate immune cell defenses, autophagy contributes an important host defense mechanism for the elimination of MRSA and other bacterial pathogens.
Concluding Remarks
S. aureus provokes strong host responses during infection but circumvents the host’s immune system by secreting an extraordinary repertoire of virulence factors. Together these factors help subvert the complement system, the activity of immune cells (phagocytosis, chemotaxis, NETs formation) or promote their killing (76, 172, 173). The selective exploitation of host cell death machineries constitutes an additional strategy that secures invasion, spread, and intra-host survival of this bacterium. S. aureus-mediated demolition of host tissues and immune cells involves all key mechanisms by which programmed host cell death can occur, including immunologically silent apoptosis and highly inflammatory signaling pathways such as pyroptosis. Several outstanding questions remain to be examined. Does S. aureus gain any advantage by provoking both non- and pro-inflammatory cell death programs? This answer may depend on the environment where the pathogen proliferates as distinct host defense arsenals may be triggered in different organ tissues. For example, deep-seated abscess formation is accompanied by the biosynthesis of apoptogenic dAdo from NETs, allowing S. aureus to selectively kill macrophages through apoptosis (37, 39). In this environment, S. aureus converts host molecules to both toxigenic and immuno-suppressive products and the infected organ fails to alert the immune system of the presence of bacteria (37, 39). On the contrary, abscess lesions in the skin elicit necroptosis, toxin-mediated activation of the NLRP3 inflammasome, and a massive recruitment of neutrophils that release pro-inflammatory cytokines such as IL-1β (103, 108, 174). Since genetically modified mice with lesions in the pyroptotic or necroptotic signaling pathway develop larger skin lesions and exhibit impaired disease outcome during bacteremia models, it appears reasonable to assume that certain cell death modes may selectively be activated by the host to limit the severity of staphylococcal infections (103, 108). In line with this model, the cell death-driven magnitude of inflammation determines the outcome of S. aureus disease and local pathology, further demonstrating that pro-inflammatory death cascades may be in favor of the mammalian host (108). If so, one wonders why S. aureus is unable to subvert pro-inflammatory host cell death modes through anti-pyroptosis or anti-necroptosis mechanisms. Presumably, the extraordinary life cycle of S. aureus requires a delicate balance between immunologically silent and inflammatory death signaling pathways in order to develop disease. Alternatively, inflammatory death signaling cascades may promote dissemination during infection or transmission to other hosts. Indeed, excessive inflammation during skin and systemic diseases is generally believed to correlate with exacerbated disease outcomes and increased mortality rates, and may therefore represent a selective infection strategy by S. aureus to establish infection (66, 108). Concomitantly, coordinated and precise perturbation of different cell death programs and cytoprotective autophagic signaling routes may help the pathogen shift from an invasive to a persistent lifestyle, thereby contributing to its global success in both healthcare facilities and the community.
Overall, S. aureus-mediated manipulation of major cell death programs, autophagy, and contributing signaling pathways substantially affects staphylococcal disease pathogenesis and clinical manifestations in many aspects. Unravelling all facets and principle mechanisms by which S. aureus modulates host cell death, along with the identification of contributing host genetic determinants, may aid the design of new therapeutic approaches to combat MRSA and other drug-resistant bacterial pathogens that exploit host cell death machineries during acute or chronic infections.
Author Contributions
VW performed the literature review and data collection, and prepared the manuscript draft and figures. DM provided revisions and comments. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all members of our laboratories for their constant support and insights and for sharing their enthusiasm for the study of host pathogen interactions. We apologize to authors whose work was either not referenced or not discussed. We are grateful for support from the National Institute of Allergy and Infectious Diseases (awards AI038897 and AI052474 to DM) and from the German Research Foundation (award WI4582/2-1 to VW).
References
1. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124(4):783–801. doi: 10.1016/j.cell.2006.02.015
2. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev (2009) 22(2):240–73. Table of Contents. doi: 10.1128/CMR.00046-08
3. Diamond G, Beckloff N, Weinberg A, Kisich KO. The roles of antimicrobial peptides in innate host defense. Curr Pharm Design (2009) 15(21):2377–92. doi: 10.2174/138161209788682325
4. Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res (2010) 20(1):34–50. doi: 10.1038/cr.2009.139
5. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol (2017) 17(3):151–64. doi: 10.1038/nri.2016.147
6. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol (2009) 7(2):99–109. doi: 10.1038/nrmicro2070
7. Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol (2015) 16(6):329–44. doi: 10.1038/nrm3999
8. Orzalli MH, Kagan JC. Apoptosis and Necroptosis as Host Defense Strategies to Prevent Viral Infection. Trends Cell Biol (2017) 27(11):800–9. doi: 10.1016/j.tcb.2017.05.007
9. Lamkanfi M, Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe (2010) 8(1):44–54. doi: 10.1016/j.chom.2010.06.007
10. Ashida H, Mimuro H, Ogawa M, Kobayashi T, Sanada T, Kim M, et al. Cell death and infection: a double-edged sword for host and pathogen survival. J Cell Biol (2011) 195(6):931–42. doi: 10.1083/jcb.201108081
11. Behar SM, Briken V. Apoptosis inhibition by intracellular bacteria and its consequence on host immunity. Curr Opin Immunol (2019) 60:103–10. doi: 10.1016/j.coi.2019.05.007
12. Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, Springer B, et al. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe (2008) 3(4):224–32. doi: 10.1016/j.chom.2008.03.003
13. Sly LM, Hingley-Wilson SM, Reiner NE, McMaster WR. Survival of Mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1. J Immunol (2003) 170(1):430–7. doi: 10.4049/jimmunol.170.1.430
14. Banga S, Gao P, Shen X, Fiscus V, Zong WX, Chen L, et al. Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc Natl Acad Sci USA (2007) 104(12):5121–6. doi: 10.1073/pnas.0611030104
15. Lowy FD. Staphylococcus aureus infections. New Engl J Med (1998) 339(8):520–32. doi: 10.1056/NEJM199808203390806
16. David MZ, Daum RS. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev (2010) 23(3):616–87. doi: 10.1128/CMR.00081-09
17. Zhang X, Hu X, Rao X. Apoptosis induced by Staphylococcus aureus toxins. Microbiol Res (2017) 205:19–24. doi: 10.1016/j.micres.2017.08.006
18. Fraunholz M, Sinha B. Intracellular Staphylococcus aureus: live-in and let die. Front Cell Infect Microbiol (2012) 2:43. doi: 10.3389/fcimb.2012.00043
19. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol (2007) 35(4):495–516. doi: 10.1080/01926230701320337
20. Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science (1997) 275(5303):1132–6. doi: 10.1126/science.275.5303.1132
21. Westphal D, Dewson G, Czabotar PE, Kluck RM. Molecular biology of Bax and Bak activation and action. Biochim Biophys Acta (2011) 1813(4):521–31. doi: 10.1016/j.bbamcr.2010.12.019
22. Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science (1997) 275(5303):1129–32. doi: 10.1126/science.275.5303.1129
23. Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell (2002) 111(3):331–42. doi: 10.1016/S0092-8674(02)01036-X
24. Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell (1996) 86(1):147–57. doi: 10.1016/S0092-8674(00)80085-9
25. Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature (1999) 399(6735):483–7. doi: 10.1038/20959
26. Abu-Hamad S, Arbel N, Calo D, Arzoine L, Israelson A, Keinan N, et al. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J Cell Sci (2009) 122(Pt 11):1906–16. doi: 10.1242/jcs.040188
27. Camara AKS, Zhou Y, Wen PC, Tajkhorshid E, Kwok WM. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front Physiol (2017) 8:460. doi: 10.3389/fphys.2017.00460
28. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell (1997) 91(4):479–89. doi: 10.1016/S0092-8674(00)80434-1
29. Cain K, Brown DG, Langlais C, Cohen GM. Caspase activation involves the formation of the aposome, a large (approximately 700 kDa) caspase-activating complex. J Biol Chem (1999) 274(32):22686–92. doi: 10.1074/jbc.274.32.22686
30. Ulett GC, Adderson EE. Regulation of Apoptosis by Gram-Positive Bacteria: Mechanistic Diversity and Consequences for Immunity. Curr Immunol Rev (2006) 2(2):119–41. doi: 10.2174/157339506776843033
31. Bantel H, Sinha B, Domschke W, Peters G, Schulze-Osthoff K, Janicke RU. alpha-Toxin is a mediator of Staphylococcus aureus-induced cell death and activates caspases via the intrinsic death pathway independently of death receptor signaling. J Cell Biol (2001) 155(4):637–48. doi: 10.1083/jcb.200105081
32. Genestier AL, Michallet MC, Prevost G, Bellot G, Chalabreysse L, Peyrol S, et al. Staphylococcus aureus Panton-Valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. J Clin Investigation (2005) 115(11):3117–27. doi: 10.1172/JCI22684
33. Haslinger B, Strangfeld K, Peters G, Schulze-Osthoff K, Sinha B. Staphylococcus aureus alpha-toxin induces apoptosis in peripheral blood mononuclear cells: role of endogenous tumour necrosis factor-alpha and the mitochondrial death pathway. Cell Microbiol (2003) 5(10):729–41. doi: 10.1046/j.1462-5822.2003.00317.x
34. Sohn MH, Kim JW, Kim WK, Jang GC, Kim KE. Staphylococcal enterotoxin B upregulates fas-mediated apoptosis of peripheral blood mononuclear cells in childhood atopic dermatitis. Scandinavian J Immunol (2003) 57(1):62–7. doi: 10.1046/j.1365-3083.2003.01183.x
35. Zhang X, Shang W, Yuan J, Hu Z, Peng H, Zhu J, et al. Positive Feedback Cycle of TNFalpha Promotes Staphylococcal Enterotoxin B-Induced THP-1 Cell Apoptosis. Front Cell Infect Microbiol (2016) 6:109. doi: 10.3389/fcimb.2016.00109
36. Imre G, Heering J, Takeda AN, Husmann M, Thiede B, Heringdorf DM, et al. Caspase-2 is an initiator caspase responsible for pore-forming toxin-mediated apoptosis. EMBO J (2012) 31(11):2615–28. doi: 10.1038/emboj.2012.93
37. Thammavongsa V, Missiakas DM, Schneewind O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science (2013) 342(6160):863–6. doi: 10.1126/science.1242255
38. Winstel V, Missiakas D, Schneewind O. Staphylococcus aureus targets the purine salvage pathway to kill phagocytes. Proc Natl Acad Sci USA (2018) 115(26):6846–51. doi: 10.1073/pnas.1805622115
39. Winstel V, Schneewind O, Missiakas D. Staphylococcus aureus Exploits the Host Apoptotic Pathway To Persist during Infection. mBio (2019) 10(6):e02270-19. doi: 10.1128/mBio.02270-19
40. Menzies BE, Kourteva I. Staphylococcus aureus alpha-toxin induces apoptosis in endothelial cells. FEMS Immunol Med Microbiol (2000) 29(1):39–45. doi: 10.1111/j.1574-695X.2000.tb01503.x
41. Prince LR, Graham KJ, Connolly J, Anwar S, Ridley R, Sabroe I, et al. Staphylococcus aureus induces eosinophil cell death mediated by alpha-hemolysin. PloS One (2012) 7(2):e31506. doi: 10.1371/journal.pone.0031506
42. Liang X, Ji Y. Involvement of alpha5beta1-integrin and TNF-alpha in Staphylococcus aureus alpha-toxin-induced death of epithelial cells. Cell Microbiol (2007) 9(7):1809–21. doi: 10.1111/j.1462-5822.2007.00917.x
43. Porichis F, Morou A, Baritaki S, Spandidos DA, Krambovitis E. Activation-induced cell death signalling in CD4+ T cells by staphylococcal enterotoxin A. Toxicol Lett (2008) 176(1):77–84. doi: 10.1016/j.toxlet.2007.10.009
44. Renno T, Hahne M, Tschopp J, MacDonald HR. Peripheral T cells undergoing superantigen-induced apoptosis in vivo express B220 and upregulate Fas and Fas ligand. J Exp Med (1996) 183(2):431–7. doi: 10.1084/jem.183.2.431
45. Ionin B, Hammamieh R, Shupp JW, Das R, Pontzer CH, Jett M. Staphylococcal enterotoxin B causes differential expression of Rnd3 and RhoA in renal proximal tubule epithelial cells while inducing actin stress fiber assembly and apoptosis. Microbial Pathogen (2008) 45(5-6):303–9. doi: 10.1016/j.micpath.2008.07.002
46. Liu Y, Chen W, Ali T, Alkasir R, Yin J, Liu G, et al. Staphylococcal enterotoxin H induced apoptosis of bovine mammary epithelial cells in vitro. Toxins (2014) 6(12):3552–67. doi: 10.3390/toxins6123552
47. Korea CG, Balsamo G, Pezzicoli A, Merakou C, Tavarini S, Bagnoli F, et al. Staphylococcal Esx proteins modulate apoptosis and release of intracellular Staphylococcus aureus during infection in epithelial cells. Infect Immun (2014) 82(10):4144–53. doi: 10.1128/IAI.01576-14
48. Berends ETM, Zheng X, Zwack EE, Menager MM, Cammer M, Shopsin B, et al. Staphylococcus aureus Impairs the Function of and Kills Human Dendritic Cells via the LukAB Toxin. mBio (2019) 10(1):e01918-18. doi: 10.1128/mBio.01918-18
49. Bu S, Xie Q, Chang W, Huo X, Chen F, Ma X. LukS-PV induces mitochondrial-mediated apoptosis and G0/G1 cell cycle arrest in human acute myeloid leukemia THP-1 cells. Int J Biochem Cell Biol (2013) 45(8):1531–7. doi: 10.1016/j.biocel.2013.05.011
50. Chi CY, Lin CC, Liao IC, Yao YC, Shen FC, Liu CC, et al. Panton-Valentine leukocidin facilitates the escape of Staphylococcus aureus from human keratinocyte endosomes and induces apoptosis. J Infect Dis (2014) 209(2):224–35. doi: 10.1093/infdis/jit445
51. Towhid ST, Nega M, Schmidt EM, Schmid E, Albrecht T, Munzer P, et al. Stimulation of platelet apoptosis by peptidoglycan from Staphylococcus aureus 113. Apoptosis An Int J Programmed Cell Death (2012) 17(9):998–1008. doi: 10.1007/s10495-012-0718-1
52. Claro T, Widaa A, O’Seaghdha M, Miajlovic H, Foster TJ, O’Brien FJ, et al. Staphylococcus aureus protein A binds to osteoblasts and triggers signals that weaken bone in osteomyelitis. PloS One (2011) 6(4):e18748. doi: 10.1371/journal.pone.0018748
53. Stelzner K, Hertlein T, Sroka A, Moldovan A, Paprotka K, Kessie D, et al. Intracellular Staphylococcus aureus employs the cysteine protease staphopain A to induce host cell death in epithelial cells. bioRxiv (2020) 2020:2020.02.10.936575. doi: 10.1101/2020.02.10.936575
54. Smagur J, Guzik K, Magiera L, Bzowska M, Gruca M, Thogersen IB, et al. A new pathway of staphylococcal pathogenesis: apoptosis-like death induced by Staphopain B in human neutrophils and monocytes. J Innate Immun (2009) 1(2):98–108. doi: 10.1159/000181014
55. Hofer MF, Newell K, Duke RC, Schlievert PM, Freed JH, Leung DY. Differential effects of staphylococcal toxic shock syndrome toxin-1 on B cell apoptosis. Proc Natl Acad Sci USA (1996) 93(11):5425–30. doi: 10.1073/pnas.93.11.5425
56. Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, et al. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PloS One (2009) 4(10):e7446. doi: 10.1371/journal.pone.0007446
57. Munoz-Planillo R, Franchi L, Miller LS, Nunez G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol (2009) 183(6):3942–8. doi: 10.4049/jimmunol.0900729
58. Soong G, Chun J, Parker D, Prince A. Staphylococcus aureus activation of caspase 1/calpain signaling mediates invasion through human keratinocytes. J Infect Dis (2012) 205(10):1571–9. doi: 10.1093/infdis/jis244
59. Ghimire L, Paudel S, Jin L, Baral P, Cai S, Jeyaseelan S. NLRP6 negatively regulates pulmonary host defense in Gram-positive bacterial infection through modulating neutrophil recruitment and function. PloS Pathogens (2018) 14(9):e1007308. doi: 10.1371/journal.ppat.1007308
60. Cohen TS, Boland ML, Boland BB, Takahashi V, Tovchigrechko A, Lee Y, et al. S. aureus Evades Macrophage Killing through NLRP3-Dependent Effects on Mitochondrial Trafficking. Cell Rep (2018) 22(9):2431–41. doi: 10.1016/j.celrep.2018.02.027
61. Hanamsagar R, Torres V, Kielian T. Inflammasome activation and IL-1beta/IL-18 processing are influenced by distinct pathways in microglia. J Neurochem (2011) 119(4):736–48. doi: 10.1111/j.1471-4159.2011.07481.x
62. Wang X, Eagen WJ, Lee JC. Orchestration of human macrophage NLRP3 inflammasome activation by Staphylococcus aureus extracellular vesicles. Proc Natl Acad Sci USA (2020) 117(6):3174–84. doi: 10.1073/pnas.1915829117
63. Perret M, Badiou C, Lina G, Burbaud S, Benito Y, Bes M, et al. Cross-talk between Staphylococcus aureus leukocidins-intoxicated macrophages and lung epithelial cells triggers chemokine secretion in an inflammasome-dependent manner. Cell Microbiol (2012) 14(7):1019–36. doi: 10.1111/j.1462-5822.2012.01772.x
64. Melehani JH, James DB, DuMont AL, Torres VJ, Duncan JA. Staphylococcus aureus Leukocidin A/B (LukAB) Kills Human Monocytes via Host NLRP3 and ASC when Extracellular, but Not Intracellular. PloS Pathogens (2015) 11(6):e1004970. doi: 10.1371/journal.ppat.1004970
65. Holzinger D, Gieldon L, Mysore V, Nippe N, Taxman DJ, Duncan JA, et al. Staphylococcus aureus Panton-Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J Leukocyte Biol (2012) 92(5):1069–81. doi: 10.1189/jlb.0112014
66. Muller S, Wolf AJ, Iliev ID, Berg BL, Underhill DM, Liu GY. Poorly Cross-Linked Peptidoglycan in MRSA Due to mecA Induction Activates the Inflammasome and Exacerbates Immunopathology. Cell Host Microbe (2015) 18(5):604–12. doi: 10.1016/j.chom.2015.10.011
67. Syed AK, Reed TJ, Clark KL, Boles BR, Kahlenberg JM. Staphlyococcus aureus phenol-soluble modulins stimulate the release of proinflammatory cytokines from keratinocytes and are required for induction of skin inflammation. Infect Immun (2015) 83(9):3428–37. doi: 10.1128/IAI.00401-15
68. Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, et al. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PloS Pathogens (2015) 11(4):e1004820. doi: 10.1371/journal.ppat.1004820
69. Essmann F, Bantel H, Totzke G, Engels IH, Sinha B, Schulze-Osthoff K, et al. Staphylococcus aureus alpha-toxin-induced cell death: predominant necrosis despite apoptotic caspase activation. Cell Death Diff (2003) 10(11):1260–72. doi: 10.1038/sj.cdd.4401301
70. Wong Fok Lung T, Monk IR, Acker KP, Mu A, Wang N, Riquelme SA, et al. Staphylococcus aureus small colony variants impair host immunity by activating host cell glycolysis and inducing necroptosis. Nat Microbiol (2020) 5(1):141–53. doi: 10.1038/s41564-019-0597-0
71. Zhou Y, Niu C, Ma B, Xue X, Li Z, Chen Z, et al. Inhibiting PSMalpha-induced neutrophil necroptosis protects mice with MRSA pneumonia by blocking the agr system. Cell Death Dis (2018) 9(3):362. doi: 10.1038/s41419-018-0398-z
72. Mestre MB, Fader CM, Sola C, Colombo MI. Alpha-hemolysin is required for the activation of the autophagic pathway in Staphylococcus aureus-infected cells. Autophagy (2010) 6(1):110–25. doi: 10.4161/auto.6.1.10698
73. Maurer K, Reyes-Robles T, Alonzo F,3, Durbin J, Torres VJ, Cadwell K. Autophagy mediates tolerance to Staphylococcus aureus alpha-toxin. Cell Host Microbe (2015) 17(4):429–40. doi: 10.1016/j.chom.2015.03.001
74. Lopez de Armentia MM, Gauron MC, Colombo MI. Staphylococcus aureus Alpha-Toxin Induces the Formation of Dynamic Tubules Labeled with LC3 within Host Cells in a Rab7 and Rab1b-Dependent Manner. Front Cell Infect Microbiol (2017) 7:431. doi: 10.3389/fcimb.2017.00431
75. Liu PF, Cheng JS, Sy CL, Huang WC, Yang HC, Gallo RL, et al. IsaB Inhibits Autophagic Flux to Promote Host Transmission of Methicillin-Resistant Staphylococcus aureus. J Invest Dermatol (2015) 135(11):2714–22. doi: 10.1038/jid.2015.254
76. Otto M. Staphylococcus aureus toxins. Curr Opin Microbiol (2014) 17:32–7. doi: 10.1016/j.mib.2013.11.004
77. Kahl BC, Goulian M, van Wamel W, Herrmann M, Simon SM, Kaplan G, et al. Staphylococcus aureus RN6390 replicates and induces apoptosis in a pulmonary epithelial cell line. Infect Immun (2000) 68(9):5385–92. doi: 10.1128/IAI.68.9.5385-5392.2000
78. Wesson CA, Deringer J, Liou LE, Bayles KW, Bohach GA, Trumble WR. Apoptosis induced by Staphylococcus aureus in epithelial cells utilizes a mechanism involving caspases 8 and 3. Infect Immun (2000) 68(5):2998–3001. doi: 10.1128/IAI.68.5.2998-3001.2000
79. Menzies BE, Kourteva I. Internalization of Staphylococcus aureus by endothelial cells induces apoptosis. Infect Immun (1998) 66(12):5994–8. doi: 10.1128/IAI.66.12.5994-5998.1998
80. Alexander EH, Rivera FA, Marriott I, Anguita J, Bost KL, Hudson MC. Staphylococcus aureus - induced tumor necrosis factor - related apoptosis - inducing ligand expression mediates apoptosis and caspase-8 activation in infected osteoblasts. BMC Microbiol (2003) 3:5. doi: 10.1186/1471-2180-3-5
81. Murai M, Sakurada J, Seki K, Shinji H, Hirota Y, Masuda S. Apoptosis observed in BALB/3T3 cells having ingested Staphylococcus aureus. Microbiol Immunol (1999) 43(7):653–61. doi: 10.1111/j.1348-0421.1999.tb02453.x
82. Tucker KA, Reilly SS, Leslie CS, Hudson MC. Intracellular Staphylococcus aureus induces apoptosis in mouse osteoblasts. FEMS Microbiol Lett (2000) 186(2):151–6. doi: 10.1111/j.1574-6968.2000.tb09096.x
83. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature (1992) 356(6372):768–74. doi: 10.1038/356768a0
84. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10(2):417–26. doi: 10.1016/S1097-2765(02)00599-3
85. Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, et al. and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature (2006) 440(7081):233–6. doi: 10.1038/nature04517
86. Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature (2006) 440(7081):228–32. doi: 10.1038/nature04515
87. Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature (2008) 452(7183):103–7. doi: 10.1038/nature06664
88. Saxena M, Yeretssian G. NOD-Like Receptors: Master Regulators of Inflammation and Cancer. Front Immunol (2014) 5:327. doi: 10.3389/fimmu.2014.00327
89. Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Diff (2007) 14(9):1590–604. doi: 10.1038/sj.cdd.4402194
90. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature (2004) 430(6996):213–8. doi: 10.1038/nature02664
91. Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T, Alnemri ES. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J Biol Chem (2001) 276(30):28309–13. doi: 10.1074/jbc.C100250200
92. Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem (2002) 277(24):21119–22. doi: 10.1074/jbc.C200179200
93. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526(7575):666–71. doi: 10.1038/nature15541
94. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
95. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535(7610):111–6. doi: 10.1038/nature18590
96. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535(7610):153–8. doi: 10.1038/nature18629
97. MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A. Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity (2001) 15(5):825–35. doi: 10.1016/S1074-7613(01)00229-1
98. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
99. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514(7521):187–92. doi: 10.1038/nature13683
100. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature (2017) 547(7661):99–103. doi: 10.1038/nature22393
101. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38(6):1142–53. doi: 10.1016/j.immuni.2013.05.016
102. Ma J, Gulbins E, Edwards MJ, Caldwell CC, Fraunholz M, Becker KA. Staphylococcus aureus alpha-Toxin Induces Inflammatory Cytokines via Lysosomal Acid Sphingomyelinase and Ceramides. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol (2017) 43(6):2170–84. doi: 10.1159/000484296
103. Miller LS, Pietras EM, Uricchio LH, Hirano K, Rao S, Lin H, et al. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J Immunol (2007) 179(10):6933–42. doi: 10.4049/jimmunol.179.10.6933
104. Perrone EE, Jung E, Breed E, Dominguez JA, Liang Z, Clark AT, et al. Mechanisms of methicillin-resistant Staphylococcus aureus pneumonia-induced intestinal epithelial apoptosis. Shock (2012) 38(1):68–75. doi: 10.1097/SHK.0b013e318259abdb
105. Singh PK, Kumar A. Mitochondria mediates caspase-dependent and independent retinal cell death in Staphylococcus aureus endophthalmitis. Cell Death Discov (2016) 2:16034. doi: 10.1038/cddiscovery.2016.34
106. Hanamsagar R, Aldrich A, Kielian T. Critical role for the AIM2 inflammasome during acute CNS bacterial infection. J Neurochem (2014) 129(4):704–11. doi: 10.1111/jnc.12669
107. Robinson KM, Ramanan K, Clay ME, McHugh KJ, Pilewski MJ, Nickolich KL, et al. The inflammasome potentiates influenza/Staphylococcus aureus superinfection in mice. JCI Insight (2018) 3(7):e97470. doi: 10.1172/jci.insight.97470
108. Kitur K, Wachtel S, Brown A, Wickersham M, Paulino F, Penaloza HF, et al. Necroptosis Promotes Staphylococcus aureus Clearance by Inhibiting Excessive Inflammatory Signaling. Cell Rep (2016) 16(8):2219–30. doi: 10.1016/j.celrep.2016.07.039
109. Krause K, Daily K, Estfanous S, Hamilton K, Badr A, Abu Khweek A, et al. Caspase-11 counteracts mitochondrial ROS-mediated clearance of Staphylococcus aureus in macrophages. EMBO Rep (2019) 20(12):e48109. doi: 10.15252/embr.201948109
110. Kebaier C, Chamberland RR, Allen IC, Gao X, Broglie PM, Hall JD, et al. Staphylococcus aureus alpha-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J Infect Dis (2012) 205(5):807–17. doi: 10.1093/infdis/jir846
111. Maher BM, Mulcahy ME, Murphy AG, Wilk M, O’Keeffe KM, Geoghegan JA, et al. Nlrp-3-driven interleukin 17 production by gammadeltaT cells controls infection outcomes during Staphylococcus aureus surgical site infection. Infect Immun (2013) 81(12):4478–89. doi: 10.1128/IAI.01026-13
112. Cao M, Chen F, Xie N, Cao MY, Chen P, Lou Q, et al. c-Jun N-terminal kinases differentially regulate TNF- and TLRs-mediated necroptosis through their kinase-dependent and -independent activities. Cell Death Dis (2018) 9(12):1140. doi: 10.1038/s41419-018-1189-2
113. Tam VC, Suen R, Treuting PM, Armando A, Lucarelli R, Gorrochotegui-Escalante N, et al. PPARalpha exacerbates necroptosis, leading to increased mortality in postinfluenza bacterial superinfection. Proc Natl Acad Sci USA (2020) 117(27):15789–98. doi: 10.1073/pnas.2006343117
114. Keller MD, Ching KL, Liang FX, Dhabaria A, Tam K, Ueberheide BM, et al. Decoy exosomes provide protection against bacterial toxins. Nature (2020) 579(7798):260–4. doi: 10.1038/s41586-020-2066-6
115. Gibson JF, Prajsnar TK, Hill CJ, Tooke AK, Serba JJ, Tonge RD, et al. Neutrophils use selective autophagy receptor Sqstm1/p62 to target Staphylococcus aureus for degradation in vivo in zebrafish. Autophagy (2020) 2020:1–10. doi: 10.1080/15548627.2020.1765521
116. Prajsnar TK, Serba JJ, Dekker BM, Gibson JF, Masud S, Fleming A, et al. The autophagic response to Staphylococcus aureus provides an intracellular niche in neutrophils. Autophagy (2020) 2020:1–15. doi: 10.1080/15548627.2020.1739443
117. Niebuhr M, Baumert K, Heratizadeh A, Satzger I, Werfel T. Impaired NLRP3 inflammasome expression and function in atopic dermatitis due to Th2 milieu. Allergy (2014) 69(8):1058–67. doi: 10.1111/all.12428
118. Zhu X, Zhang K, Lu K, Shi T, Shen S, Chen X, et al. Inhibition of pyroptosis attenuates Staphylococcus aureus-induced bone injury in traumatic osteomyelitis. Ann Trans Med (2019) 7(8):170. doi: 10.21037/atm.2019.03.40
119. Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity (2012) 36(3):464–76. doi: 10.1016/j.immuni.2012.02.001
120. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol (2014) 15(2):135–47. doi: 10.1038/nrm3737
121. He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell (2009) 137(6):1100–11. doi: 10.1016/j.cell.2009.05.021
122. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol (2000) 1(6):489–95. doi: 10.1038/82732
123. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell (2012) 148(1-2):213–27. doi: 10.1016/j.cell.2011.11.031
124. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science (2009) 325(5938):332–6. doi: 10.1126/science.1172308
125. Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol (1988) 141(8):2629–34.
126. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell (2003) 114(2):181–90. doi: 10.1016/S0092-8674(03)00521-X
127. Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, et al. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med (1998) 188(5):919–30. doi: 10.1084/jem.188.5.919
128. Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell (2008) 30(6):689–700. doi: 10.1016/j.molcel.2008.05.014
129. Csomos RA, Brady GF, Duckett CS. Enhanced cytoprotective effects of the inhibitor of apoptosis protein cellular IAP1 through stabilization with TRAF2. J Biol Chem (2009) 284(31):20531–9. doi: 10.1074/jbc.M109.029983
130. Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell (1995) 83(7):1243–52. doi: 10.1016/0092-8674(95)90149-3
131. Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature (2004) 430(7000):694–9. doi: 10.1038/nature02794
132. Xu G, Tan X, Wang H, Sun W, Shi Y, Burlingame S, et al. Ubiquitin-specific peptidase 21 inhibits tumor necrosis factor alpha-induced nuclear factor kappaB activation via binding to and deubiquitinating receptor-interacting protein 1. J Biol Chem (2010) 285(2):969–78. doi: 10.1074/jbc.M109.042689
133. Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell (2008) 135(7):1311–23. doi: 10.1016/j.cell.2008.10.044
134. Wright A, Reiley WW, Chang M, Jin W, Lee AJ, Zhang M, et al. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev Cell (2007) 13(5):705–16. doi: 10.1016/j.devcel.2007.09.007
135. O’Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol (2011) 13(12):1437–42. doi: 10.1038/ncb2362
136. Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal (2007) 19(10):2056–67. doi: 10.1016/j.cellsig.2007.05.016
137. Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev (1999) 13(19):2514–26. doi: 10.1101/gad.13.19.2514
138. Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature (2011) 471(7338):363–7. doi: 10.1038/nature09852
139. Pop C, Oberst A, Drag M, Van Raam BJ, Riedl SJ, Green DR, et al. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J (2011) 433(3):447–57. doi: 10.1042/BJ20101738
140. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol (2010) 11(10):700–14. doi: 10.1038/nrm2970
141. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity (2013) 39(3):443–53. doi: 10.1016/j.immuni.2013.06.018
142. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell (2014) 54(1):133–46. doi: 10.1016/j.molcel.2014.03.003
143. Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol (2014) 16(1):55–65. doi: 10.1038/ncb2883
144. He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA (2011) 108(50):20054–9. doi: 10.1073/pnas.1116302108
145. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem (2013) 288(43):31268–79. doi: 10.1074/jbc.M113.462341
146. Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell (2011) 43(3):432–48. doi: 10.1016/j.molcel.2011.06.006
147. Wen SH, Lin LN, Wu HJ, Yu L, Lin L, Zhu LL, et al. TNF-alpha increases Staphylococcus aureus-induced death of human alveolar epithelial cell line A549 associated with RIP3-mediated necroptosis. Life Sci (2018) 195:81–6. doi: 10.1016/j.lfs.2018.01.008
148. Greenlee-Wacker MC, Rigby KM, Kobayashi SD, Porter AR, DeLeo FR, Nauseef WM. Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J Immunol (2014) 192(10):4709–17. doi: 10.4049/jimmunol.1302692
149. Kobayashi SD, Braughton KR, Palazzolo-Ballance AM, Kennedy AD, Sampaio E, Kristosturyan E, et al. Rapid neutrophil destruction following phagocytosis of Staphylococcus aureus. J Innate Immun (2010) 2(6):560–75. doi: 10.1159/000317134
150. Gonzalez-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, et al. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PloS Pathogens (2015) 11(12):e1005337. doi: 10.1371/journal.ppat.1005337
151. Greenlee-Wacker MC, Kremserova S, Nauseef WM. Lysis of human neutrophils by community-associated methicillin-resistant Staphylococcus aureus. Blood (2017) 129(24):3237–44. doi: 10.1182/blood-2017-02-766253
152. Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M, et al. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat Rev Microbiol (2006) 4(4):295–305. doi: 10.1038/nrmicro1384
153. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol (2018) 19(6):349–64. doi: 10.1038/s41580-018-0003-4
154. Yu L, Chen Y, Tooze SA. Autophagy pathway: Cellular and molecular mechanisms. Autophagy (2018) 14(2):207–15. doi: 10.1080/15548627.2017.1378838
155. Khandia R, Dadar M, Munjal A, Dhama K, Karthik K, Tiwari R, et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells (2019) 8(7):674. doi: 10.3390/cells8070674
156. Bonam SR, Wang F, Muller S. Lysosomes as a therapeutic target. Nat Rev Drug Discov (2019) 18(12):923–48. doi: 10.1038/s41573-019-0036-1
157. Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell (2000) 103(2):253–62. doi: 10.1016/S0092-8674(00)00117-3
158. Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol (2010) 22(2):132–9. doi: 10.1016/j.ceb.2009.12.004
159. Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol (2010) 191(1):155–68. doi: 10.1083/jcb.201002100
160. Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell (2014) 55(2):238–52. doi: 10.1016/j.molcel.2014.05.021
161. Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Diff (2019) 26(4):605–16. doi: 10.1038/s41418-018-0252-y
162. Jiao Y, Sun J. Bacterial Manipulation of Autophagic Responses in Infection and Inflammation. Front Immunol (2019) 10:2821. doi: 10.3389/fimmu.2019.02821
163. Kimmey JM, Stallings CL. Bacterial Pathogens versus Autophagy: Implications for Therapeutic Interventions. Trends Mol Med (2016) 22(12):1060–76. doi: 10.1016/j.molmed.2016.10.008
164. Schnaith A, Kashkar H, Leggio SA, Addicks K, Kronke M, Krut O. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J Biol Chem (2007) 282(4):2695–706. doi: 10.1074/jbc.M609784200
165. Neumann Y, Bruns SA, Rohde M, Prajsnar TK, Foster SJ, Schmitz I. Intracellular Staphylococcus aureus eludes selective autophagy by activating a host cell kinase. Autophagy (2016) 12(11):2069–84. doi: 10.1080/15548627.2016.1226732
166. Wang H, Zhou Y, Zhu Q, Zang H, Cai J, Wang J, et al. Staphylococcus aureus induces autophagy in bovine mammary epithelial cells and the formation of autophagosomes facilitates intracellular replication of Staph. aureus. J Dairy Sci (2019) 102(9):8264–72. doi: 10.3168/jds.2019-16414
167. Keil E, Hocker R, Schuster M, Essmann F, Ueffing N, Hoffman B, et al. Phosphorylation of Atg5 by the Gadd45beta-MEKK4-p38 pathway inhibits autophagy. Cell Death Diff (2013) 20(2):321–32. doi: 10.1038/cdd.2012.129
168. Geng N, Wang X, Yu X, Wang R, Zhu Y, Zhang M, et al. Staphylococcus aureus Avoids Autophagy Clearance of Bovine Mammary Epithelial Cells by Impairing Lysosomal Function. Front Immunol (2020) 11:746. doi: 10.3389/fimmu.2020.00746
169. O’Keeffe KM, Wilk MM, Leech JM, Murphy AG, Laabei M, Monk IR, et al. Manipulation of Autophagy in Phagocytes Facilitates Staphylococcus aureus Bloodstream Infection. Infect Immun (2015) 83(9):3445–57. doi: 10.1128/IAI.00358-15
170. Soong G, Paulino F, Wachtel S, Parker D, Wickersham M, Zhang D, et al. Methicillin-resistant Staphylococcus aureus adaptation to human keratinocytes. mBio (2015) 6(2):e00289-15. doi: 10.1128/mBio.00289-15
171. Bravo-Santano N, Ellis JK, Mateos LM, Calle Y, Keun HC, Behrends V, et al. Intracellular Staphylococcus aureus Modulates Host Central Carbon Metabolism To Activate Autophagy. mSphere (2018) 3(4):e00374-18. doi: 10.1128/mSphere.00374-18
172. Thammavongsa V, Kim HK, Missiakas D, Schneewind O. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol (2015) 13(9):529–43. doi: 10.1038/nrmicro3521
173. Spaan AN, Surewaard BG, Nijland R, van Strijp JA. Neutrophils versus Staphylococcus aureus: a biological tug of war. Annu Rev Microbiol (2013) 67:629–50. doi: 10.1146/annurev-micro-092412-155746
Keywords: Staphylococcus aureus, host cell death, persistence, infection, abscess, blood stream infection
Citation: Missiakas D and Winstel V (2021) Selective Host Cell Death by Staphylococcus aureus: A Strategy for Bacterial Persistence. Front. Immunol. 11:621733. doi: 10.3389/fimmu.2020.621733
Received: 26 October 2020; Accepted: 03 December 2020;
Published: 21 January 2021.
Edited by:
Fabio Bagnoli, GlaxoSmithKline, ItalyReviewed by:
Bernd Lepenies, University of Veterinary Medicine Hannover, GermanySrinivasa Reddy Bonam, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2021 Missiakas and Winstel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Volker Winstel, winstel.volker@mh-hannover.de